Современные
представления о строении органических соединений. Основы стереохимии органических
соединений. Ассиметрический атом углерода. Хиральность. Проекционные формулы
Фишера.
Теория химического строения А.М. Бутлерова
В 1861 году А.М. Бутлеровым была предложена теория
химического строения органических соединений, которая состоит из следующих основных
положений.
1) В молекулах веществ существует строгая
последовательность химического связывания атомов, которая называется химическим
строением.
2) Химические свойства вещества определяются природой
элементарных составных частей, их количеством и химическим строением.
3) Если у веществ с одинаковым составом и молекулярной
массой различное строение, то возникает явление изомерии.
4) Так как в конкретных реакциях изменяются только
некоторые части молекулы, то исследование строения продукта помогает определить
строение исходной молекулы.
5) Химическая природа (реакционная способность) отдельных
атомов в молекуле меняется в зависимости от окружения, т.е. от того, с какими
атомами других элементов они соединены.
Теория Бутлерова дает принципиальную возможность
познания геометрии молекулы (микроскопических свойств) через познание
химических свойств (макроскопических свойств). Основные положения теории строение
сохраняют свое значение до сих пор.
Электронные
теории химической связи.
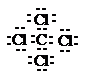
Электронное строение
органических соединений изображают с помощью электронных формул Льюиса. В них с
помощью точек указывают положение всех валентных электронов: электронов
химических связей и неподеленных пар электронов. При этом считают, что
неподеленные пары электронов составляют часть внешней оболочки только одного
атома, а электроны, участвующие в образовании ковалентной связи, являются
частью внешней оболочки обоих атомов. Например, в приведенной ниже формуле
Льюиса для тетрахлорметана все атомы имеют октет электронов.
Для каждого атома в структуре Льюиса определяют
формальный заряд. При этом полагают, что атому принадлежат все неподеленные
электроны и половина электронов ковалентных связей. Избыток электронов, принадлежащих атому в
молекуле по сравнению со свободным атомом, обусловливает отрицательный заряд, а
недостаток - положительный заряд. Сумма формальных зарядов всех атомов дает
заряд частицы в целом.

Основные принципы квантовой органической химии.

Современные теории ковалентной связи основаны на представлениях
квантовой механики. Согласно принципам квантовой механики состояние электрона в
атоме определяется волновой функцией, которую называют атомной орбиталью. Образование
химической связи между атомами рассматривается как результат взаимодействия
двух орбиталей, на каждой из которых находится по одному электрону. При этом происходит
образование молекулярных орбиталей (МО). Из двух атомных орбиталей образуются
две молекулярные орбитали, одна из которых (связывающая)
имеет более низкую энергию, а другая (разрыхляющая)
– более высокую энергию, чем исходные АО.
Электроны связи занимают более низкую по энергии связывающую орбиталь,
таким образом, взаимодействие орбиталей приводит к выигрышу в энергии.
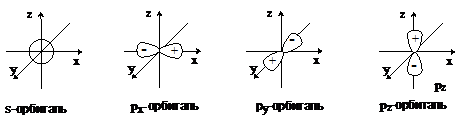
В зависимости от типа комбинирующихся атомных орбиталей образуются разные
типы МО. Определяющую роль в этом играют симметрия и узловые свойства орбиталей.
Атомные s-орбитали имеют симметрию шара и не имеют узловых поверхностей,
проходящих через центр атома. Атомные p-орбитали
имеют цилиндрическую симметрию и три состояния px, py и pz. Каждая p-орбиталь
имеет узловую плоскость, проходящую через центр атома и перпендикулярную
соответственно оси x, y или z.
Узловая поверхность – это место, где вероятность нахождения электрона
равна нулю, а волновая функция меняет знак. Чем больше узлов, тем выше энергия
орбитали. Таким образом, p-орбиталь
состоит из двух частей, в которых знаки волновых функций противоположны.
При рассмотрении электронного строения многоатомных молекул необходимо
использовать такой набор орбиталей, при котором достигается их максимальное
перекрывание. В связи с этим водится понятие гибридизации орбиталей.
Атом углерода в возбужденном состоянии содержит четыре неспаренных электрона на
внешнем энергетическом уровне и способен образовать четыре ковалентных связи.
В образовании связей участвуют гибридные орбитали.
Первое валентное состояние – sp3-гибридизация. В результате гибридизации с участием одной s и трех p
орбиталей атома углерода образуются четыре эквивалентные sp3-гибридные
орбитали, направленные к вершинам тетраэдра под углами 109,5о:

В состоянии sp3-гибридизации атом углерода
образует четыре s-связи с четырьмя заместителями и имеет тетраэдричекую конфигурацию с
валентными углами, равными или близкими 109,5о:
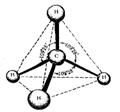
Метан
Второе валентное состояние – sp2-гибридизация. В результате гибридизации с участием одной s- и двух
p-орбиталей атома углерода образуются три эквивалентные sp2-гибридные
орбитали, лежащие в одной плоскости под углами 120о, а не
участвующая в гибридизации p-орбиталь расположена перпендикулярно плоскости
гибридных орбиталей.
В состоянии sp2-гибридизации атом углерода
образует три s-связи за счет гибридных орбиталей и одну p-связь за счет не участвующей в гибридизации p-орбитали
и имеет три заместителя.
Третье валентное состояние углерода – sp-гибридизация. В результате гибридизации с участием одной s- и одной
p–орбитали образуются две эквивалентные sp-гибридные орбитали, лежащие под
углом 1800, а не участвующие в гибридизации p-орбитали расположены
перпендикулярно плоскости гибридных орбиталей и друг другу. В состоянии
sp-гибридизации атом углерода образует две s-связи за счет гибридных орбиталей и две p-связи за счет не участвующих в гибридизации
p-орбиталей и имеет два заместителя:
Ацетилен
Основы
стереохимии.
Стереохимия – часть химии, посвященная изучению пространственного строения
молекул и влияния этого строения на физические и химические свойства вещества,
на направление и скорость их реакций.
Конформации
(поворотная изомерия).
Переход от простейшего органического углеводорода – метана,
к его ближайшему гомологу – этану ставит проблемы пространственного строения,
для решения которых недостаточно знать рассмотренные ранее параметры. Не меняя
ни валентных углов, ни длин связей, можно представить себе множество геометрических
форм молекулы этана, отличающихся друг от друга взаимным поворотом углеродных
тетраэдров вокруг соединяющей их связи С-С. В результате такого вращения
возникают поворотные изомеры (конформеры).
Энергия различных конформеров неодинакова, но энергетический барьер,
разделяющий различные поворотные изомеры, для большинства органических
соединений невелик. Поэтому при обычных условиях, как правило, нельзя
зафиксировать молекулы в одной строго определенной конформации. Обычно в
равновесии сосуществуют несколько легко переходящих друг в друга поворотных
форм.
Рассмотрим способы графического изображения
конформаций и их номенклатуру. Для молекулы этана можно предвидеть
существование двух максимально различающихся по энергии конформаций. Они изображены
ниже в виде перспективных проекций (1)
("лесопильные козлы"), боковых проекций (2)
и формул Ньюмена.
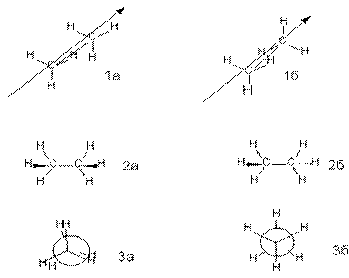
Изображенную слева конформацию называют заслоненной.
Это название напоминает о том, что атомы водорода обеих СН3-групп
находятся друг против друга. Заслоненная конформация имеет повышенную внутреннюю
энергию, и поэтому невыгодна. Конформацию, изображенную справа, называют заторможенной, подразумевая, что свободное вращение вокруг
связи С-С "тормозится" в этом положении, т.е. молекула существует преимущественно
в этой конформации.
С усложнением молекулы число возможных конформаций
возрастает. Так, для н-бутана можно
изобразить уже шесть конформаций, отличающихся взаимным расположением СН3-групп,
т.е. поворотом вокруг центральной связи С-С. Ниже конформации н-бутана
изображены в виде проекций Ньюмена. Изображенные слева (заслоненные)
конформации энергетически невыгодны, практически реализуются лишь заторможенные.
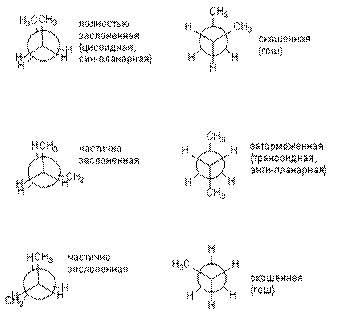
Различные заслоненные и заторможенные конформации бутана
неодинаковы по энергии. Соответствующие энергии всех конформаций, образующихся
при вращении вокруг центральной С-С связи.
Итак, конформации – это различные пространственные
формы молекулы, имеющей определенную конфигурацию. Конформерами являются стереоизомерные
структуры, соответствующие энергетическим минимумам на диаграмме потенциальной
энергии, находящиеся в подвижном равновесии и способные к взаимопревращению
путем вращения вокруг простых связей.
Иногда барьер таких превращений становится достаточно
высоким, чтобы разделить стереоизомерные формы (пример - оптически активные дифенилы).
В таких случаях говорят уже не о конформерах, а о реально существующих стереоизомерах.
Важное следствие жесткости двойной связи (отсутствия
вращения вокруг нее) – существование геометрических изомеров.
Самые распространенные из них – это цис-,транс-изомеры
соединений этиленового ряда, содержащих у ненасыщенных атомов неодинаковые
заместители. Простейшим примером могут служить изомеры бутена-2.

Геометрические изомеры имеют одинаковое химическое
строение, различаясь по пространственному расположению атомов, т.е. по конфигурации. Это различие и создает разницу в физических
(а также химических свойствах). Геометрические изомеры, в отличие от конформеров,
могут быть выделены в чистом виде и существуют как индивидуальные устойчивые вещества.
Для их взаимного превращения необходима энергия порядка 125 – 170 кДж/моль,
которую можно сообщить нагреванием или
облучением.
В простейших случаях номенклатура геометрических
изомеров не представляет затруднений: цис-формами называют геометрические
изомеры, у которых одинаковые заместители лежат по одну сторону от плоскости
пи-связи, транс-изомеры имеют одинаковые заместители на разных сторонах
от плоскости пи-связи. В более сложных случаях применяется Z,E-номенклатура.
Ее главный принцип: для обозначения конфигурации указывают цис- (Z, от немецкого Zusammen - вместе) или транс- (Е, от немецкого Entgegen - напротив) расположение старших заместителей при двойной связи.
В Z,E-системе старшими считаются заместители с большим
атомным номером. Если атомы, непосредственно связанные с ненасыщенными углеродами,
одинаковы, то переходят ко "второму слою", в случае необходимости - к
"третьему слою" и т.д.
3. Оптическая изомерия (энантиомерия).
Среди органических соединений встречаются вещества,
способные вращать плоскость поляризации света. Это явление называют оптической
активностью, а соответствующие вещества – оптически активными.
Оптически активные вещества встречаются в виде пар оптических
антиподов - изомеров, физические и химические свойства которых в
обычных условиях одинаковы, за исключением одного – знака вращения плоскости
поляризации. (Если один из оптических антиподов имеет, например, удельное вращение
+20о, то другой - удельное вращение -20о).
Оптическая изомерия появляется тогда, когда в молекуле
присутствует асимметрический атом углерода.
Так называют атом углерода, связанный с четырьмя различными заместителями.
Возможны два тетраэдрических расположения заместителей вокруг асимметрического
атома. Обе пространственные формы нельзя совместить никаким вращением; одна из
них является зеркальным изображением другой.
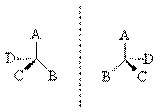
Рассмотренный вид изомерии называют оптической
изомерией, зеркальной изомерией или энантиомерией. Обе зеркальные формы составляют пару оптических
антиподов или энантиомеров.
Для условного изображения асимметрического атома на
плоскости пользуются проекционными формулами Э.Фишера.
Их получают, проецируя на плоскость атомы, с которыми связан асимметрический
атом. При этом сам асимметрический атом, как правило, опускают, сохраняя лишь
перекрещивающиеся линии и символы заместителей. Чтобы помнить о пространственном
расположении заместителей, часто сохраняют в проекционных формулах прерывистую
вертикальную линию (верхний и нижний заместитель удалены за плоскость чертежа),
однако часто этого не делают. левой модели на предыдущем рисунке:
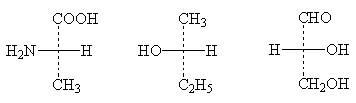
Приведем несколько примеров проекционных формул:
(+)-аланин (-)2-бутанол (+)-глицериновый
альдегид
При названиях веществ приведены их знаки вращения. Это значит, например,
что левовращающий антипод бутанола-2 имеет пространственную
конфигурацию, выражаемую именно приведенной выше формулой, а ее зеркальное
изображение отвечает правовращающему бутанолу-2. Определение
конфигурации оптических антиподов проводится экспериментально.
В принципе, каждый оптический антипод может быть
изображен двенадцатью (!) различными проекционными формулами - в зависимости от
того, как расположена модель при построении проекции, с какой стороны мы
смотрим на нее. Чтобы стандартизировать проекционные формулы, введены
определенные правила их написания. Так, главную функцию, если она находится в
конце цепи, принято ставить наверху, главную цепь изображать вертикально.
Для того чтобы сопоставлять "нестандартно"
написанные проекционные формулы, надо знать следующие правила преобразования
проекционных формул.Для того чтобы сопоставлять "нестандартно"
написанные проекционные формулы, надо знать следующие правила преобразования
проекционных формул.
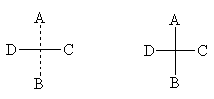
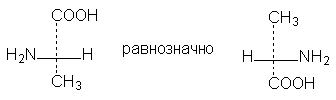
1. Формулы можно вращать в плоскости чертежа на 180о,
не меняя их стереохимического смысла:
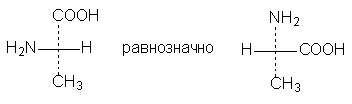
2. Две (или любое четное число) перестановки
заместителей у одного асимметрического атома не меняют стереохимического смысла
формулы:
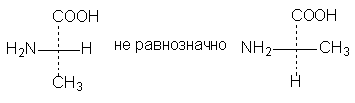
3. Одна (или любое нечетное число) перестановок
заместителей у асимметрического центра приводит к формуле оптического антипода:
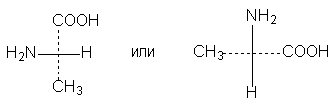
4. Поворот в плоскости чертежа на 90о
превращает формулу в антиподную, если только при этом одновременно не изменить
условие расположения заместителей относительно плоскости чертежа, т.е. не
считать, что теперь боковые заместители находятся за плоскостью чертежа, а
верхний и нижний - перед ней. Если пользоваться формулой с пунктиром, то изменившаяся
ориентация пунктира прямо напомнит об этом:
5. Вместо перестановок проекционные формулы можно
преобразовывать путем вращения любых трех заместителей по часовой стрелке или
против нее; четвертый заместитель при этом положения не меняет (такая операция
эквивалентна двум перестановкам):
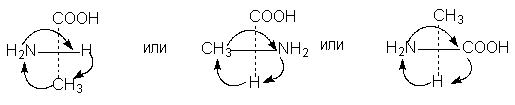
6. Проекционные формулы нельзя выводить из плоскости
чертежа.
Рацематы.
Если в формуле вещества есть асимметрический атом, это
отнюдь не означает, что такое вещество будет обладать оптической активностью.
Если асимметрический центр возникает в ходе обычной реакции (замещение в группе
СН2, присоединение по двойной связи и т.п.), то вероятность создания
обеих антиподных конфигураций одинакова. Поэтому, несмотря на асимметрию каждой
отдельной молекулы, получающееся вещество оказывается оптически неактивным.
Такого рода оптически неактивные модификации, состоящие из равного количества
обоих антиподов, называются рацематами.
Другие типы оптически активных веществ.
В этом разделе перечислены некоторые другие классы
органических соединений, также обладающих оптической активностью (т.е.
существующие в виде пар оптических антиподов).
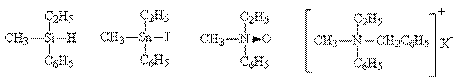
Атом углерода не обладает монополией на создание хиральных
центров в молекулах органических соединений. Центром хиральности могут быть
также атомы кремния, олова, четырехковалентного азота в четвертичных аммониевых
солях и окисях третичных аминов:
В этих соединениях центр асимметрии имеет
тетраэдрическую конфигурацию, как и асимметрический атом углерода. Существуют,
однако, и соединения с иной пространственной структурой хирального центра.
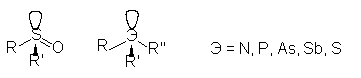
Пирамидальную конфигурацию имеют хиральные центры, образованные атомами
трехвалентного азота, фосфора, мышьяка, сурьмы, серы. В принципе, центр
асимметрии можно считать тетраэдрическим, если в качестве четвертого заместителя
принять неподеленную электронную пару гетероатома:
Оптическая активность может возникать и без
хирального центра, за счет хиральности структуры всей молекулы в целом (молекулярная хиральность или молекулярная асимметрия).
Наиболее характерными примерами являются наличие хиральной
оси либо хиральной плоскости.
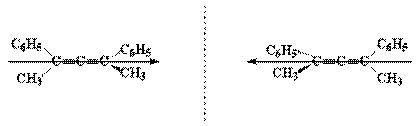
Хиральная ось возникает, например, в алленах,
содержащих различные заместители при sp2-гибридных
углеродных атомах. Легко видеть, что приведенные ниже соединения являются
несовместимыми зеркальными изображениями, а значит оптическими антиподами:
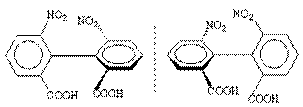
Другой класс соединений, имеющих хиральную ось -
оптически активные бифенилы, которые имеют в орто-положениях объемистые заместители, затрудняющие свободное
вращение вокруг С-С связи, соединяющей бензольные ядра:
Хиральная плоскость
характеризуется тем, что у нее можно различить "верх" и
"низ", а также "правую" и "левую" стороны.
Примером соединений с хиральной плоскостью могут служить оптически активный транс-циклооктен и оптически активное
производное ферроцена:
Диастереомерия.
Соединения с несколькими асимметрическими атомами
обладают важными особенностями, отличающими их от рассмотренных ранее более простых
оптически активных веществ с одним центром асимметрии.
Допустим, что в молекуле некоего вещества имеются два
асимметрических атома; обозначим их условно А и Б. Легко видеть, что возможны молекулы
со следующими комбинациями:
|
|
((-) |
((-) |
|
((-) |
((+) |
|
Молекула 1 |
FА |
<Б |
Молекула 3 |
АА |
ББ |
|
|
((+) |
((+) |
|
((+) |
((-) |
|
Молекула 2 |
АА |
ББ |
Молекула 4 |
АА |
ББ |
Молекулы 1 и 2 представляют собой пару оптических
антиподов; то же самое относится и к паре молекул 3 и 4. Если же сравнивать
друг с другом молекулы из разных пар антиподов - 1 и 3, 1 и 4, 2 и 3, 2 и 4, то
мы увидим, что перечисленные пары не являются оптическими антиподами: конфигурация
одного асимметрического атома у них совпадает, конфигурация другого - не
совпадает. Это пары диастереомеров, т.е. пространственных
изомеров, не составляющих друг с другом оптических антиподов.
Диастереомеры отличаются друг от друга не только
оптическим вращением, но и всеми другими физическими константами: у них разные
температуры плавления и кипения, разные растворимости и др. Различия в свойствах
диастереомеров зачастую ничуть не меньше, чем различия в свойствах между
структурными изомерами.
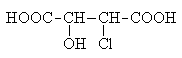
Примером соединения рассматриваемого типа может
случить хлоряблочная кислота
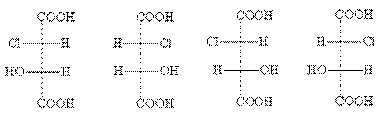
Ее стереоизомерные формы имеют следующие проекционные
формулы:
эритро-формы трео-формы
Названия эритро-
и трео- происходят от названий
углеводов эритрозы и треозы. Эти названия употребляют для указания взаимного
положения заместителей у соединений с двумя асимметрическими атомами: эритро-изомерами называют те, у которых два одинаковых
боковых заместителя стоят в стандартной проекционной формуле на одной стороне (справа
или слева); трео-изомеры имеют одинаковые
боковые заместители на разных сторонах проекционной формулы. Два эритро-изомера представляют собой пару
оптических антиподов, при их смешении образуется рацемат. Парой оптических
изомеров являются и трео-формы; они
тоже дают при смешении рацемат, отличающийся по свойствам от рацемата эритро-формы. Таким образом, всего
существуют четыре оптически активных изомера хлоряблочной кислоты и два
рацемата.
При дальнейшем росте числа асимметрических центров
число пространственных изомеров возрастает, причем каждый новый асимметрический
центр вдвое увеличивает число изомеров. Оно определяется формулой 2n,
где n - число асимметрических центров.
Число стереоизомеров может уменьшаться из-за частичной
симметрии, появляющейся в некоторых структурах. Примером может служить винная
кислота, у которой число индивидуальных стереоизомеров сокращается до трех. Их
проекционные формулы:
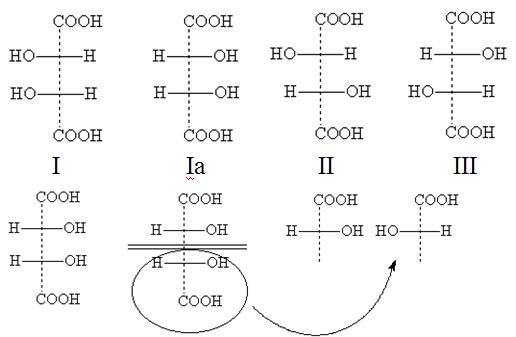
Формула I идентична с формулой Iа, так как
превращается в нее при повороте на 180о в плоскости чертежа и,
следовательно, не изображает нового стереоизомера. Это оптически неактивная
модификация называется мезо-форма.
Мезо-формы имеются у всех оптически
активных веществ с несколькими одинаковыми (т.е. связанными с одинаковыми
заместителями) асимметрическими центрами. Проекционные формулы мезо-форм всегда можно узнать по тому,
что их можно разделить горизонтальной линией на две половины, которые по записи
на бумаге формально идентичны, в действительности же зеркальны:
Формулы II и III изображают оптические антиподы винной
кислоты; при их смешении образуется оптически неактивный рацемат - виноградная
кислота.
Номенклатура оптических изомеров.

Самая простая, наиболее старая, однако и ныне еще
употребляемая система номенклатуры оптических антиподов, основана на сравнении
проекционной формулы называемого антипода с проекционной формулой некоего
стандартного вещества, выбранного в качестве "ключа". Так, для a-оксикислот и a-аминокислот ключом является верхняя часть их проекционной
формулы (в стандартной записи):
L-оксикислоты (Х = ОН) D-оксикислоты
(Х = ОН)
L-аминокислоты
(Х = NH2) D-аминокислоты (Х = NH2)
Конфигурацию всех a-оксикислот, имеющих в стандартно написанной проекционной
формуле Фишера гидроксильную группу слева, обозначают знаком L; если же гидроксил расположен в
проекционной формуле справа - знаком D
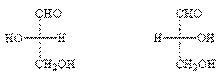
Ключом для обозначения конфигурации сахаров служит
глицериновый альдегид:
L-(-)-глицериновый альдегид D-(+)-глицериновый
альдегид
В молекулах сахаров обозначение D- или L- относится к
конфигурации нижнего асимметрического
центра.
Система D-,L-обозначений имеет существенные
недостатки: во-первых, обозначение D-
или L- указывает конфигурацию только
одного асимметрического атома, во-вторых, для некоторых соединений получаются
разные обозначения, в зависимости от того, взят ли в качестве ключа глицериновый
альдегид или оксикислотный ключ, например:
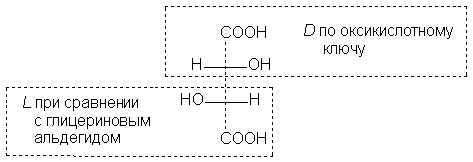
Эти недостатки системы ключей ограничивают ее применение
в настоящее время тремя классами оптически активных веществ: сахарами, аминокислотами
и оксикислотами. На общее же применение рассчитана R,S-система
Кана, Ингольда и Прелога.
Для определения R- или S-конфигурации оптического
антипода необходимо расположить тетраэдр заместителей вокруг асимметрического
углеродного атома таким образом, чтобы младший заместитель (обычно это водород)
имел направление "от наблюдателя". Тогда, если движение при переходе
по кругу трех остальных заместителей от старшего к среднему по старшинству и
затем к самому младшему происходит против часовой стрелки - это S-изомер (ассоциируется с таким же движением руки при написании
буквы S), если по часовой стрелке - это R-изомер (ассоциируется с движением
руки при написании буквы R).
Для определения старшинства заместителей у
асимметрического атома используются правила подсчета атомных номеров, уже
рассмотренные нами в связи с Z,E-номенклатурой геометрических изомеров.
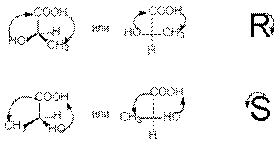
Для выбора R,S-обозначений по проекционной формуле
необходимо путем четного числа перестановок (не изменяющих, как мы знаем,
стереохимического смысла формулы) расположить заместители так, чтобы младший из
них (обычно водород) оказался внизу проекционной формулы. Тогда старшинство
остальных трех заместителей, падающее по часовой стрелке, соответствует
обозначению R, против часовой стрелки - обозначению S:
5. Методы получения стереоизомеров
Получение чистых стереоизомеров – важная задача, так
как, как правило, только одна из стереоизомерных форм является биологически
активной. Между тем, в обычных условиях образуются, как правило, смеси стереоизомеров
- диастереомеров или оптических антиподов. Для получения чистых стереоизомерных
форм эти смеси.