Галогенопроизводные
углеводородов. Реакции нуклеофильного замещения и элиминирования, их механизмы;
влияние структуры субстрата и растворителя.
Галогенпроизводные углеводородов можно рассматривать
как результат замещения однако или более атомов водорода в углеводороде на
атомы галогенов. В зависимости от характера связи С-Hal различают следующие
типы галогенуглеводородов:
1. Галогенпроизводные со связью С(sp3)-Hal
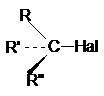
где R, R1, R11 - атом водорода
или алкильной радикал.
2. Галогенпроизводные со связью С(sp2)-Hal

3. Галогенпроизводные со связью С(sp)-Hal
R-CºC-Hal
Галогенпроизводные
типа С(sp3)-Hal.
Методы получения.
1. Заместительное галогенирование.
Объектом заместительного галогенирования могут быть алканы. В общем
виде реакции заместительного галогенирования алканов выражаются уравнением:
RH + Hal2
® RHal + HНal
Таким способом можно получить фтор-, бром- и
хлоралканы. Подробно об особенностях этих реакций – см. химические свойства алканов.
Аллильное хлорирование алканов можно осуществить при
высоких температурах (400-500оС) в паровой фазе
t
СН2 = СН – СH3 + Cl2 ® CH2 =
CH – CH2Cl + HCl
Для аллильного бромирования алканов в качестве
реагента используют бромсукцинимид
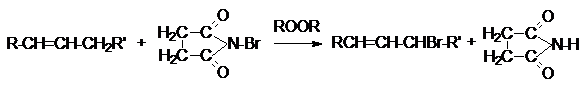
Хлорирование и бромирование боковых цепей алкиларенов можно осуществлять
в условиях инициирования свободнорадикальных реакций (химические инициаторы,
фотоинициирование), причем наиболее реакционноспособным оказывается a-положения боковой цепи:
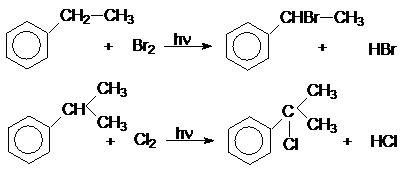
2. Присоединительное галогенирование алкенов и
алкинов.
Возможности этого метода можно проиллюстрировать следующими реакциями:
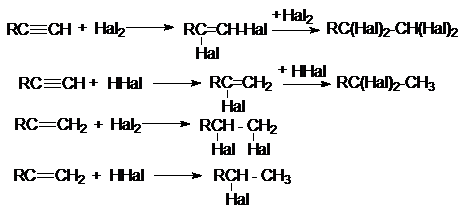
Особенности приведенных реакций подробно обсуждались в
разделах, посвященных химическим свойствам алкенов и алкинов. Здесь лишь
отметим, что при реализации этих реакций в промышленности с целью форсирования
процессов используют катализаторы типа кислот Льюиса (FeCl3, SnCl4,
SbCl3 и др.)
3. Реакции замещения гидроксильных групп на галоген в
спиртах.
К таким реакциям относятся:
а) взаимодействие галогеноводородов со спиртами
ZnCl2
ROH + HCl ––––® RCl + H2O
Важным фактором эффективности этих реакций является
высокая кислотность среды, обеспечивающая стабилизацию уходящей гидроксильной
группы:
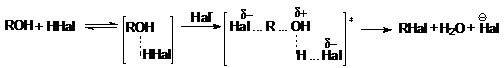
Каталитическое действие хлорида цинка связано с
образованием более сильной протонной кислоты за счет донорно-акцепторного взаимодействия:
ZnCl2 +
HCl « H2[ZnCl4]
которая в последующем выполняет каталитическую
функцию. В связи с важностью поддержания кислотности среды в этих реакциях в
качестве гидрогалогенирующих реагентов можно использовать комбинации
соль-кислота, например, KBr + H2SO4.
Иногда в качестве реагентов используют смеси,
продуцирующие in situ необходимый галогеноводород:
2P + 3J2
+ 6H2O ® 6HJ + 2H3PO3
б) Взаимодействие галогенангидридов кислот (PСl3,
PВr3, PСl5, SOCl2, PJ3 и др.) со
спиртами:
PCl3 +
3ROH ® 3rcl + H3PO3
PCl5 +
5ROH ® 5rcl + H3PO4
+ H2O
SOCl2 +
ROH ® RCl + SO2 + HCl
SF4 +
2KOH ® 2RF + SO2 + 2HF
в) Взаимодействие галогенангидридов кислот (PСl5,
PBr5, SF4) c альдегидами и
кетонами
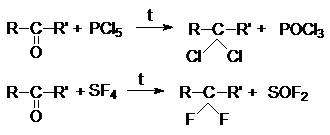
где R’=H, R, Ar.
4. Замещение галогена на галоген.
Замещение более легкого галогена в галогенуглеводороде
более тяжелым
RCl + KBr ® RВr + KCl
обеспечивается проведением реакции при умеренных
температурах и избытке галогенирующего реагента в неполярных апротонных
растворителях.
Замещение более тяжелого галогена более легким
RJ + KCl ® RCl + KJ
обеспечивается проведением реакции при повышенных
температурах и избытке галогенирующего реагента в высокополярной среде в
присутствии катализатров - кислот Люиса, например, Сu+, Ag+
и пр.
5. Галогенметилирование аренов
cat
ArH + CH2O + HНal –––® ArCH2Hal + H2O
t
Физические
свойства и строение.
Галогенпроизводные углеводородов являются бесцветными
газами или жидкостями со своеобразным сладковатым запахом. В воде нерастворимы
и в большинстве случаев тяжелее ее. Некоторые полигалогенпроизводные образуют
бесцветные кристаллы, полииодсоединения имеют желтую окраску.
Температура кипения возрастает при увеличении атомной
массы галогена и числа атомов галогена и углерода. Исключением являются
полифторалканы, температура кипения которых уменьшается при увеличении числа
атомов фтора в молекуле полифторалкана. При переходе от фторпроизводных к подпроизводным
увеличивается их поляризуемость, что соответствует ряду поляризуемости атомов:
J > Br > Cl > F
Энергия связи С-Hal меняется в ряду
D(C – F) > D(C –
Cl) > D(C – Br) > D(C – J)
Моногалогенпроизводные имеют значительные дипольные
моменты, что свидетельствует о полярности связи С-Hal. Это связано с более
высокой электроотрицательностью атомов галогенов по сравнению с углеродными
атомами:
![]()
Неподеленные пары электронов на атомах галогенов
придают молекуле галогенуглеводородов слабые электронодонорные свойства,
которые увеличиваются в ряду
RJ > RBr >
RCl > RF
Химические
свойства.
Химическое поведение галогенуглеводородов определяется
такими факторами как энергия связи С–Hal, полярность этой связи и ее поляризуемость.
Так, относительная слабость связей С–Cl, C–Br и СJ обусловливает их предпочтительное
гомолитическое расщепление по сравнению со связями С–С и С–Н. В то же время
полярность связей С–Hal и их более
высокая поляризуемость по сравнению со связями С–С и С–Н является предпосылкой
для их гетеролитического расщепления. Приводимые ниже реакции являются иллюстрацией
этих положений.
1. Замещение галогена на водород.
Восстановление галогенпроизводных до углеводородов
осуществляется водородом в присутствии обычных катализаторов гидрирования (Ni,
Pt, Pd):
cat
RCH2Hal + H2 ––––––––® RCH3 + HНal
150-300oC
Реакция имеет гомолитический характер, связанный с
предварительной сорбцией реагентов на поверхности катализатора, причем водород
претерпевает диссоциативную адсорбцию с вовлечением адсорбированных атомов водорода
в реакцию восстановления
H2 + S «2[H·]адс
RCH2Hal
+ S1 « [RCH2Cl]адс
[RCH2Cl]адс + [H·]адс ® [RCH2·]адс + [HCl]адс
[RCH2·]адс + [H·]адс ® [RCH3]адс
[RCH3]адс « RCH3 + S
[HCl]адс « HCl
где S – поверхность катализатора.
Реакция (1) имеет важное практическое значение как
метод переработки галогенорганических отходов в промышленном органическом
синтезе. В отличие от другого метода обезвреживания галогенорганических
отходов, сжигания, этот метод является ресурсосберегающим и экологически безопасным,
так как при его реализации регенерируется углеводородная составляющая исходного
сырья и исключается образование высокотоксичных полихлордибензодиоксинов и полихлордибензофуранов.
Другой метод замещения галогена на водород –
взаимодействие галогенпроизводных углеводородов с иодоводородной кислотой при
нагревании
t
RCH2Hal
+ 2HI ® RCH3 + HHal + I2
Метод имеет препаративное значение.
2. Взаимодействие с металлами.
а) реакция
димеризации (синтез Вюрца)
2RHal + Na ® R–R + 2NaHal
Механизм этой реакции может быть представлен следующей
последовательностью стадий:
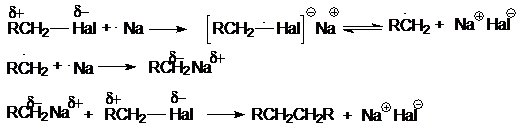
Такой характер взаимодействия является выражением
природы реагентов. Натрий как очень активный металл легко отдает свой электрон
молекуле галогенуглеводорода, которая выступает в роли слабого электрофила.
Электрофильные свойства молекулы R–Hal вытекают из полярности связи С–Hal,
обусловливающей дефицит электронов на атоме углерода. Образующийся интермедиат
распадается на алкильный радикал и натриевую соль галогена, причем прочная ионная
связь в последней обусловливает легкость этого распада. Аналогично можно
объяснить энергетическую выгодность последующих реакций механизма.
Реакция имеет препаративное значение.
б)
Взаимодействие с магнием (реакция Гриньяра).
[ROR]
RCH2Hal
+ Mg –––––® RCH2MgHal
Механизм этой реакции в части ее инициирования подобен
механизму реакции Вюрца
![]()
Реактивы Гриньяра имеют важнейшее значение в
препаративной органической химии, поскольку открывают возможности получения
широкого разнообразия органических соединений на основе взаимодействия этих
реактивов с различными реагентами.
3. Реакции нуклеофильного замещения.
Реакции нуклеофильного замещения – наиболее типичный
круг реакций, в которых галогенуглеводороды выступают в качестве субстратов.
Результатом этих реакций является замещение галогена на другой атом или группу,
которые либо непосредственно выступают в роли нуклеофильного реагента, либо
входят в его состав в качестве
фрагмента.
Наиболее типичными реакциями нуклеофильного замещения
галогеналканов и других галогенпроизводных являются:
а) реакции гидролиза
RHal + H2O ® ROH + HHal
RHal + NaOH ® ROH + NaHal
б) реакции образования простых эфиров (реакция
Вильямсона)
RHal + R`ONa ® ROR` + NaHal
в) синтез сложных эфиров
R1Hal +
RCOONa ® RCOOR1 + NaHal
г) аммонолиз
RHal + 2NH3
® RNH2 + NH4Hal
д) синтез нитросоединений и нитритов
RHal + NaNO2
![]() ® RNO2 + NaHal
® RNO2 + NaHal
RHal + NaNO2
 ®RONO + NaHal
®RONO + NaHal
е) синтез нитрилов и изонитрилов
RHal + NaCN ![]() ® RCN + NaHal
® RCN + NaHal
RHal + NaCN ® RNC + NaHal
ж) синтез тиолов и сульфидов
R–Hal + NaSH ® RSH + NaHal
R–Hal + R1SNa
® RSR1 + NaHal
2R–Hal + Na2S
® RSR + 2NaHal
з) синтез фосфорорганических соединений
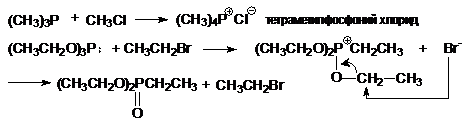
(CH3)2PH
+ CH3CH2Br + NaOH ® (CH3)2PCH2CH3
+ NaBr + H2O
диметилэтилфосфин
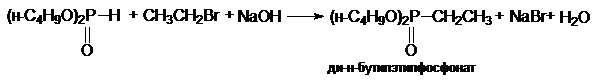
и) синтез углеводородов на основе Mg-органических
соединений
R – MgHal + R’Hal ® R–R’ + MgHal2
к) замещение галогена на галоген
RX + NaY ® RY + NaY
Из приведенных реакций можно видеть, что реакции
нуклеофильного замещения могут служить методом образования связей С–С, С–О,
С–S, C–N, C–P и C–Hal.
Закономерности
реакций нуклеофильного замещения.
Реакции некаталитического нуклеофильного замещения
могут протекать в зависимости от условий, характера реакционной среды,
структуры субстрата и природы нуклеофильного реагента по одностадийному или
двухстадийному механизмам.
Одностадийный процесс представляет собой элементарную реакцию SN2-замещения, где индекс 2 указывает на бимолекулярность реакции:
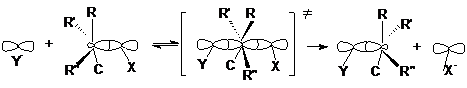
или более схематично
:Y- + RX « ![]() ¹® RY + X:-
¹® RY + X:-
Можно видеть, что нуклеофил своей неподеленной парой
электронов атакует углеродный атом в молекуле субстрата со стороны, противоположной
замещаемой группе. При образовании активированного комплекса атакуемый атом углерода
переходит из sp3-состояния в sp2-состояние. В результате
углеродный атом оказывается в одной плоскости с группами R, R’ и R”, а нуклеофил
и замещаемая группа связываются с ним за счет его р-орбитали. Распад активированного
комплекса в продукт реакции сопровождается обратным переходом углеродного атома
в sp3-состояние и обращением конфигурации исходного соединения,
которое вызвано присоединением группы Y со стороны, противоположной разорвавшейся
связи С-Х. Поэтому если объектом бимолекулярного нуклеофильного замещения
является хиральный атом углерода, то в результате реакции происходит обращение
оптической активности соединения. Скорость бимолекулярной реакции SN2
выражается уравнением
r = k[RX][Y-]
При двухстадийном нуклеофильном замещении разрыв связи
С-Х и образование связи С-Y происходит не одновременно. Первая элементарная
стадия этой реакции – мономолекулярный гетеролитический разрыв связи С-Х с образованием
карбкатиона
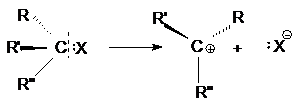
При этом центральный атом углерода переходит в
состояние sp2-гибридизации и карбкатион приобретает плоское
строение. Поэтому он с равной вероятностью атакуется нуклеофилом как со стороны
отщепившейся группы, так и с противоположной стороны.
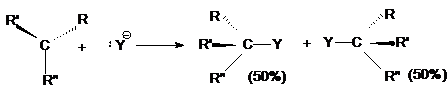
Поэтому, если объектом нуклеофильной атаки является
хиральный атом углерода, то в результате реакции оптически активного субстрата
образуется рацемическая смесь.
Первая стадия – мономолекулярная стадия гетеролиза -
является скорость-определяющей. В связи с этим двухстадийный процесс нуклеофильного
замещения обозначают символом SN1, где цифра 1 означает мономолекулярность
скорость-определяющей стадии.
Скорость реакций SN1 в соответствии с
характером скорость-определяющей стадии выражается уравнением
r = k[RX]
В действительности механизм рассматриваемых реакций
нуклеофильного замещения является более сложным.
В настоящее время общепринято, что разрыв связи С-Х
существенно облегчается или вообще становится возможным лишь за счет
взаимодействия замещаемой группы Х с электрофилами. Образование
донорно-акцепторной связи I или водородной связи II
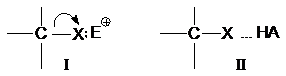
ведет к предварительной поляризации связи С-Х и
облегчает ее последующий разрыв. Замещение некоторых групп (-OH, -OR, -SH, -SR,
-NH2, -NHR, - NR2) вообще невозможно без их сильного
взаимодействия с электрофилами. Например, замещение гидроксильной группы спирта
при действии только бромид-аниона невозможно, но эту же реакцию легко
осуществить в кислой среде. Предварительное протонирование гидроксильной группы
меняет ее характер: в качестве уходящей группы выступает стабильная молекула
воды, что делает энергетически выгодным гетеролиз связи С-ОН и облегчает атаку
аниона брома
ROH + HBr « RO+H2
+ Br– « [Brd- ... R ... Od-H2]¹ ® Rbr + H2O
Аналогичным образом осуществляется превращение простых
эфиров, меркантанов и аминов.
Кроме сильных протонных кислот (НСl, H2SO4)
катализаторами реакций нуклеофильного замещения являются кислота Льюиса (Ag+,
Hg2+, HgCl, HgCl2, SnCl4, AlCl3,
SbCl5, Cu2Cl2, BF3 и др.). Их
действие зависит от природы отщепляемой группы. Например, протонные кислоты
существенно ускоряют замещение О-, S- и N-содержащих групп и мало влияют на
скорость замещения галогенов, поскольку последние являются слабыми основаниями
и плохо протонируются. Атомы галогенов дают, однако, прочные
донорно-акцепторные комплексы с некоторыми кислотами Льюиса, и их образование
облегчает разрыв связи С-Hal при последующей реакции замещения. Так, аллилхлорид
медленно гидролизуется водой, но в присутствии хлорида меди (I) легко дает аллиловый
спирт
![]() CH2=CH–CH2
® Cl : Cu2Cl2
+ H2O ® CH2=CH–CH2O+H2
+ Cu2Cl3 ® CH2=CH-CH2OH
+ HCl + Cu2Cl2
CH2=CH–CH2
® Cl : Cu2Cl2
+ H2O ® CH2=CH–CH2O+H2
+ Cu2Cl3 ® CH2=CH-CH2OH
+ HCl + Cu2Cl2
При этом может произойти изменение механизма SN2
в некаталитической реакции на SN1 - в каталитической:
медл. U
RCl + Cu2Cl2
¨ R+ + Cu2Cl3
R+ + Y-
®RY
Cu2Cl3–
® Cu2Cl2 + Cl–
Ослабление связи С-Х и уменьшение энергии переходного
состояния в реакциях нуклеофильного замещения происходит и при образовании
водородных связей между уходящей группой и протонодонорным агентом.
Так, скорость реакции метанолиза трифенилхлорметана
(C6H5)3CCl
+ CH3OH ® (C6H5)3COCH3
+ HCl
описывается кинетическим уравнение третьего порядка:
r = k[(C6H5)CCl][CH3OH]2
Это согласуется с механизмом, согласно которому
замещение хлора происходит только при условии его специфической сольватации
другой молекулой спирта:
(C6H5)3C-Cl
+ CH3OH « (C6H5)3C–Cl¼HOCH3
Образующийся ассоциат дает далее продукт замещения под
действием второй молекулы нуклеофила - спирта
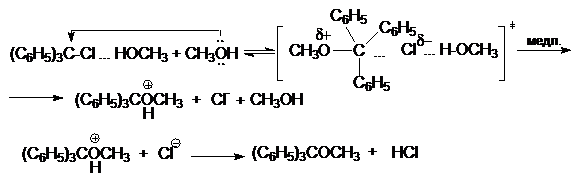
Противоположный эффект наблюдается при специфической
сольватации нуклеофила, поскольку в результате образования водородных связей
основность последнего утрачивается, что понижает его нуклеофильное сродство к
положительно заряженному реакционному центру. При этом отрицательно заряженные
нуклеофилы сольватируются сильнее нейтральных молекул или групп. Так, например,
в реакции
CH3J + Br– ® CH3Br + J–
преобладающее влияние имеет сольватация бромид-атома,
а не атома иода в молекуле СH3J. Это связано с большей электроотрицательностью
брома по сравнению с иодом, что обусловливает большую прочность водородной
связи с Br– по сравнению с отщепляющимся J–. В результате
скорость этой реакции в метаноле в десятки тысяч раз меньше, чем в апротонных
растворителях, где эффект специфической сольватации отсутствует.
Сольватация существенно влияет не только на общую скорость
реакции, но и на соотношение механизмов SN1 и SN2. При прочих
равных условиях первый из них тем более вероятен, чем выше полярность растворителя
и его способность к специфической сольватации уходящей группы и чем выше кислотность
катализатора. Такая роль катализаторов связана с выделением энергии
сольватации, компенсирующей энергетические затраты на гетеролиз С-Х-связи.
В действительности SN1 и SN2-реакции
не отделены друг от друга резкой гранью, между ними имеется область так
называемых пограничных механизмов. При “чистом” SN2-механизме с
отрицательно заряженным нуклеофилом реакционный центр в переходном состоянии
имеет частичный отрицательный заряд, поскольку образование новой связи С-Y
опережает разрыв старой связи С-Х. Если на эту реакцию начинают действовать
факторы, благоприятствующие SN1-механизму (температура, сильносольватирующая
среда, катализаторы – электрофилы) разрыв старой связи начинает опережать
образование новой; реакционный центр в переходном состоянии приобретает положительный
заряд и в пределе возникает ситуация, когда старая связь практически разорвана,
а новая не образовалась, что соответствует чистому механизму SN1.
Переход от механизма SN2 к механизму SN1 иллюстрируется следующей схемой:
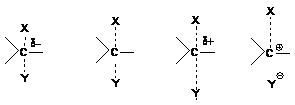
Влияние строения реагентов на реакции нуклеофильного
замещения. Влияние
структуры радикала R субстрата.
Успех реакции SN2 определяется
эффективностью атаки нуклеофила на положительно заряженный реакционный центр субстрата.
При этом, чем выше этот положительный заряд, тем эффективнее взаимодействие.
Поэтому электронодонорные заместители в радикале R, понижая положительный заряд
на реакционном центре, замедляет нуклеофильную атаку. В то же время увеличению
объема R затрудняет подход нуклеофила к реакционному центру. Совместное
действие индуктивного и объемного эффектов определяет ряд реакционных
способностей субстратов в реакциях нуклеофильного замещения:
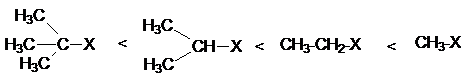
Введение электроакцепторных заместителей в радикал R
приводит к увеличению положительного заряда на реакционном центре субстрата,
что делает нуклеофильную атаку более эффективной. Так, аллилхлорид гидролизуется
более эффективно, чем хлорпропан из-за активирующего действия отрицательного
индуктивного эффекта винильной группы:
CH2 = CH ¬ CH2Cl CH3CH2 ® CH2Cl
В реакциях SN1 скорость-определяющей стадией
является образованием карбкатиона. В соответствии с постулатом Хэммонда высокая
эндотермичность этой стадии обусловливает высокую степень разрыва связей в ее
переходном состоянии
[Rd+............Xd–]¹
Поэтому структура фрагмента R в переходном состоянии
приближается к структуре свободного карбкатиона и все факторы, стабилизирующие
карбкатионы, будут стабилизировать переходное состояние а, следовательно, приводить
к ускорению реакции.
Факторами стабилизации карбкатиона и, соответственно,
переходного состояния гетеролиза связи С-Х являются увеличение объема заместителей
при реакционном центре и их электронодонорные свойства. Так, ряд реакционной
способности соединений R-X в SN1-реакциях
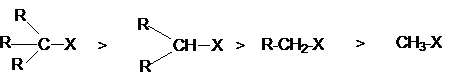
определяются совместным вкладом в стабилизацию
соответствующих этому ряду карбкатионов
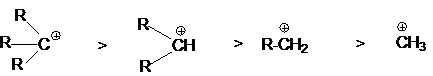
положительного индуктивного эффекта алкильных групп и
увеличением объема алкильной группы за счет ее удлинения и разветвления.
Активирующее действие +М-эффекта на скорость SN1-реакций
можно проиллюстрировать на примере гидролиза этоксиметилбромида и
2-бромпропана. Первое соединение обладает намного большей реакционной
способностью из-за существенной стабилизации карбкатиона
![]()
![]()
CH3CH2OCH2 « CH3CH2O=CH2
Противоположное влияние индуктивного и объемного
эффектов на реакции SN1 и SN2 может быть проиллюстрировано
типичной зависимостью скорости реакций нуклеофильного замещения от числа
алкильных заместителей при реакционном центре

Можно видеть, что в области SN2-механизма
электронодонорный и объемный эффект алкильных групп замедляет реакции, в
области SN1 – ускоряет. Между этими предельными случаями находится
область пограничных механизмов, когда факторы SN1 и SN2
реакций конкурируют между собой.
Влияние уходящей группы.
При прочих одинаковых условиях нуклеофильное замещение
как по SN1- так и по SN2-механизму должно протекать тем
быстрее, чем ниже энергия гетеролитического разрыва связи С-Х. Понижение этой
энергии во многом связано со стабилизацией образующегося аниона за счет его
лучшей поляризуемости (в случае галогенид-анионов) или эффектов сопряжения (для
эфиров сульфокислот и диалкилсульфенов)
В то же время у спиртов и эфиров в уходящих группах –ОН
и –OR отсутствуют структурные возможности для стабилизации, поэтому
их отщепление эффективно при действии кислотного катализа (см. выше).
Стабилизация уходящей группы гораздо рельефнее влияет
на ускорение SN1-реакций по сравнению с реакциями SN2.
Это связано с большей степенью разрыва связей в скорость-определяющей стадии SN1-реакции.
Поэтому чем больше способность к стабилизации у уходящей группы, тем больше
вероятность SN1-механизма.
Влияние нуклеофильного реагента.
В случае SN1-реакций нуклеофильный реагент
не участвует в скорость-определяющей стадии, поэтому его сила и концентрация не
оказывают влияния на скорость этих реакций.
В случае SN2-механизма нуклеофильный
реагент непосредственно участвует в реакции. Влияние его силы и концентрации
суммируется в кинетическом уравнении (1)
Можно видеть, что скорость этих реакций линейно
зависит от концентрации нуклеофильного реагента. Влияние силы нуклеофильного
реагента отражается в значении константы скорости k. Силу нуклеофильного
реагента определяется термином нуклеофильность. Нуклеофильность является
результатом вклада двух факторов: основности и поляризуемости. Связь нуклеофильности
с основностью определяется аналогией нуклеофильных реакций с протолитическими
реакциями и фактически отражает сродство нуклеофила к положительно заряженному
реакционному центру. В тех случаях, когда другие факторы, влияющие на нуклеофильность,
остаются постоянными, наблюдается корреляция между константой скорости реакции
SN2-замещения (нуклеофильностью) и константой основности нуклеофильного
реагента (основностью). Количественно эта зависимость выражается уравнением
Бренстеда
k = G × KBb или lgk = lgG + blgKB
где k -константа скорости, KB - константа
основности нуклеофила, G и b -эмпирические коэффициенты. Чем больше b, тем быстрее возрастает скорость с увеличением
основности нуклеофила. По мере перехода от SN2 к SN1-механизму
завязывание новой связи происходит на все большем расстоянии и величина b уменьшается. В пределе, когда реакция протекает по SN1-механизму,
b=0.
Другим фактором нуклеофильности является
поляризуемость. В ионах большого размера (HS–, J–, Br–) или при наличии в анионе кратных связей (CN–,
NO2–) электроны легко смещаются в направлении
положительно заряженного реакционного центра. Благодаря этому поляризованная
нуклеофильная частица завязывает с ним связь на большем расстоянии и реагирует
значительно быстрее нуклеофилов с близкой или даже большей основностью, но не
способных к поляризации. Например, усредненная относительная нуклеофильность легко
поляризуемых ионов С6H5S– в 470 раз выше для
ионов С2H5O–, тогда как основность последних
(рКВ=–2) на много порядков больше, чем для иона С6H5S–
(рКВ=–4,6). Высокой нуклеофильностью обладают также ионы НS–
и CN–, молекулы аммиака и аминов. В результате условия синтеза
меркаптанов, сульфидов, аминов, нитрилов нуклеофильным замещением хлоралканов
нередко оказываются даже более мягкими, чем условия их гидролиза щелочами.
Эдвардс предложил следующие уравнения для
количественной оценки нуклеофильности
ЕN = 3,6 lg![]() + 0,0624(pKa+1,74)
+ 0,0624(pKa+1,74)
где Ri - рефракция (поляризуемость)
нуклеофила, рКа -кислотность кислоты, сопряженной
основанию-нуклеофилу. Стандартом служит вода, для которой нуклеофильность
принята равной нулю.
Для аминов нуклеофильность изменяется в ряду: R2NH»RNH2>>NH3 При прочих равных
условиях, чем сильнее нуклеофильность реагента, тем более вероятно протекание
реакции по SN2-механизму. Наоборот, со слабыми нуклеофилами более
быстрой может оказаться реакция SN1. Такая зависимость с обращением механизмов может быть
продемонстрирована графиком зависимости скорости реакции от нуклеофильности
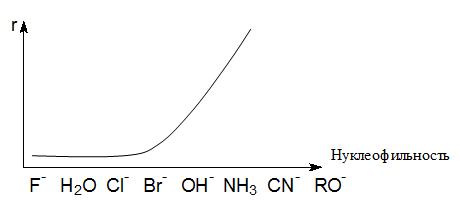
Анализ влияния природы нуклеофила и уходящей группы на
скорость реакций нуклеофильного замещения показывает, что высокая
поляризуемость увеличивает нуклеофильность частицы и одновременно делает ее
хорошей уходящей группой. Это позволяет использовать такие частицы в качестве
нуклеофильных катализаторов. Например, анион иода сильно ускоряет относительно
медленные реакции гидролиза хлоралканов водой. Механизм нуклеофильного катализа
этой реакции состоит в предварительном замещении хлорид-аниона иодом и
последующем гидролизе образовавшегося иодалкана с образованием спирта и регенерацией
J–:
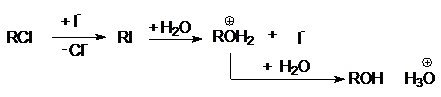
Конкуренция реакций нуклеофильного замещения
При проведении реакций нуклеофильного замещения в
реакционных смесях могут присутствовать несколько нуклеофилов. В этих случаях
имеет место конкуренция нескольких нуклеофильных реакций:
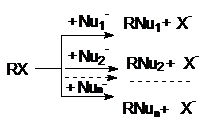
Из схемы конкуренции видно, что увеличение выхода
любого из продуктов достигается увеличением избытка нуклеофила, ответственного
за его образование. Так, при реализации щелочного гидролиза хлоралканов
протекают следующие реакции
RCl + OH–
® ROH + Cl–
ROH + OH– RO–
+ H2O
RCl + RO– ® ROR + Cl–
Можно видеть, что соотношение скоростей основной
реакции (3) и побочной реакции образования простого эфира (5) определяется
конкурентным взаимодействием
![]() ROH +OH-
ROH +OH-
![]() RCl
RCl
ROR +RO-
и увеличение выхода основного продукта, спирта, может
быть обеспечено увеличением избытка гидроксил-аниона. Поскольку соотношение ![]() определяется в соответствие
с равновесием (4) равенством
определяется в соответствие
с равновесием (4) равенством
![]()
факторами увеличения выхода спирта являются увеличение
избытка воды и снижение конверсии хлоралкана (что равносильно понижению
концентрации спирта).
Некоторые нуклеофилы обладают двумя реакционными
центрами и поэтому дают разные продукты замещения. При этом преимущественное
образование продукта замещения по тому или иному реакционному центру определяется
правилом
Корнблюма: Если реакция
протекает по SN2-механизму, то нуклеофил реагирует с субстратом
своим более нуклеофильным (поляризуемым) центром. Если реакция протекает по SN1-механизму,
то нуклеофил реагирует с субстратом своим более электроотрицательным основным
центром. Примером ионов с двойственной реакционной способностью (амбидентных
ионов) являются цианид-анион СºN и
нитрит-анион
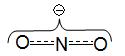
Эти анионы в зависимости от механизма реакции,
определяемого условиями их проведения и структурой субстрата, могут давать следующие
продукты
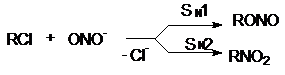
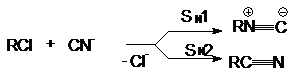
Подобная ситуация имеет место при обмене галогенов.
Реакция SN2 всегда протекает в сторону замещения более электроотрицательного
атома на более нуклеофильный (поляризуемый) (F и Cl на Br и J). Однако при SN1-замещении
образуется продукт с более электроотрицательным заместителем (например, RF из
RСl). Для изменения механизма используют соли или катализаторы с сильно электрофильным
катионом (AgF, HgF2, SbF3, SbF5):
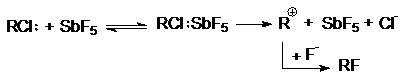
Реакции нуклеофильного отщепления.
Эти реакции называют еще реакциями элиминирования (англ.
elimination). Их можно классифицировать по взаимному расположению отщепляемых
групп (1,1-, 1,2-, 1,3-элиминирование и т.д.).
Наибольшее практическое значение имеют реакции
1,2-отщепления, ведущие к образованию ненасыщенных веществ (процессы дегидрогалогенирования,
дегидратации и др.):
RCH2CH2Cl
RCH=CH2
Cl2CH-CH2Cl
Cl2C=CH2
RCH2CH2OSO3H
RCH=CH2
RCH2-CH2-OH
R-CH=CH2
а также отдельные реакции 1,3 и 1,4-отщепления с
образованием гетероциклических соединений
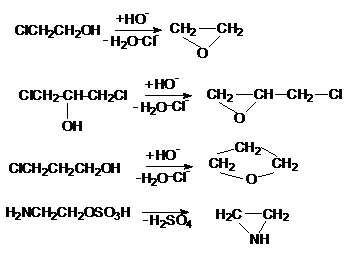
Механизм
реакций отщепления.
В зависимости от условий реакции и строения
реагирующих веществ 1,2-отщепления может протекать по разным механизмам. По очередности
разрыва связей С–Х и С–Н они подразделяются на механизмы Е1, Е2 и Е1св,
где цифры 1 и 2 – показывают молекулярность скорость-определяющей стадии,
индекс “св” означает сопряженное основание (англ. conjugated basе).
Реакция Е1 обычно конкурирует с SN1 и имеет
с ним общую скорость-определяющую стадию гетеролиза связи С-Х
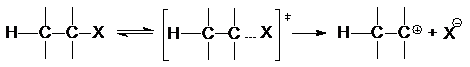
Образовавшийся карбкатион взаимодействует далее с
каким-либо основанием или растворителем, выполняющим функцию основания, с
отщеплением протона и образованием алкена:
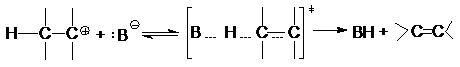
В соответствии с характером лимитирующей стадии
протеканию Е1 благоприятствуют те же факторы, что и реакции SN1, а
скорость реакции выражается кинетическим уравнением
R=k [H-C- C-X]
подобным уравнению скорости SN1 – реакции.
Как указывалось выше, к этим факторам относятся
разветвление алкильной группы и наличие электронодонорных заместителей при
реакционном центре субстрата, уменьшение энергии связи С–Х, наличие растворителя
и катализаторов, способных к электрофильному взаимодействию с Х. Типичный
пример представляет дегидратация спиртов, катализируемая протонными или апротонными
кислотами, которые стабилизируя уходящую группу, способствуют ее эффективному
отщеплению и образованию карбкатиона:
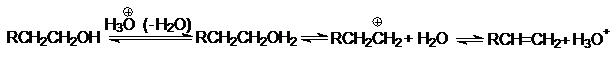
Таким образом, протонированный спирт претерпевает
мономолекулярное расщепление, определяя Е1 – характер реакции.
Поскольку стабильность ионов карбония возрастает в
ряду
трет. R+ > втор. R+ >
перв. R+
то дегидратация наиболее легко протекает у третичных
спиртов, в то время как для первичных спиртов требуются наиболее жесткие
условия.
Если в субстрате отсутствуют структурные возможности
для стабилизации карбкатиона, а реагент представляет собой сильное основание,
то реакция отщепления протекает по механизму Е2. В этом случае разрыв связей
С-Н и С-Х происходит синхронно в процессе бимолекулярной элементарной реакции
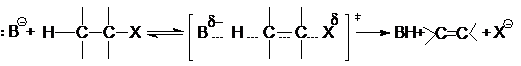
![]() и скорость такой реакции описывается кинетическим
уравнением второго порядка
и скорость такой реакции описывается кинетическим
уравнением второго порядка
r=k[B-][H-C–C-X]
подобным уравнению для реакции SN2.
По этому механизму протекают реакции расщепления
н-хлоралканов, эфиров серной кислоты и др.
Реакция Е2 ускоряется электроноакцепторными группами,
повышающими кислотность отщепляемого атома водорода. К таким группам относятся
СF3, NO2, SO2, CN, C=O, наличие которых при
том же углеродном атоме, что и отщепляемый водород, существенно ускоряет
реакцию. Примером может служить катализируемая основаниями дегидратация b-кетоспиртов
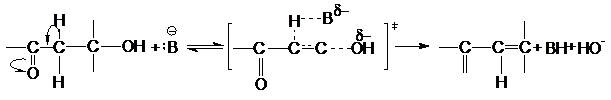
![]()
Если протон в субстрате обладает достаточно высокой
кислотностью, а группа Х отщепляется трудно, реакция протекает по механизму Е1св,
когда происходит предварительное отщепление протона по быстрой равновесной реакции
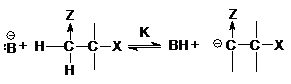
а группа Х отщепляется в следующей медленной
мономолекулярной стадии распада карбкатиона
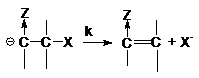
Роль электроноакцепторной группы Z заключается в поляризации отщепляемого протона и
стабилизации промежуточного аниона.
Скорость реакции в соответствии с характером
лимитирующей стадии выражается уравнением
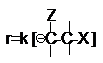
Концентрация карбаниона определяется равновесием (3)
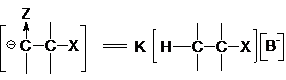
Подставляя выражение (6) в уравнение (5) приходим к
кинетическому уравнению реакции Е1св
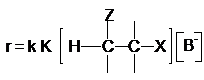 (7)
(7)
Из уравнений (2) и (7) видно, что механизмы Е2 и Е1св
кинетически неразличимы. Различие между ними можно выявить на основе анализа
активационных параметров брутто-реакций. Так, константа скорости реакции SN2
как константа истинной бимолекулярной реакции будет характеризоваться отрицательными
энтропиями активации, тогда как брутто-константа реакции Е1св должна
характеризоваться положительной энтропией активации.
Примером Е1св-реакции является
1,3-отщепление НСl от хлоргидринов с образованием оксиранов
Cl-CH2CH2OH
+ HO- « Cl-CH2CH2O-
+ H2O
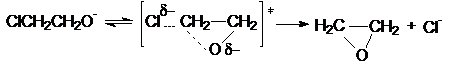
Направление
отщепления.
Если атом углерода, несущий отщепляемую группу Х,
имеет два или три соседних углеродных атома, содержащих С-Н–связи, возможно
образование нескольких изомерных алкенов. Направление отщепления в этом случае
определяется правилом Зайцева, согласно которому водород предпочтительно
отщепляется от наименее гидрированного атома углерода с образованием разветвленного
алкена. Например, при щелочном дегидробромировании 3-бром-2,3-диметилпентана доминирующим
продуктом реакции будет 2,3-диметилпентен-2
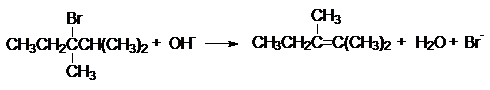
В основе такого направления отщепления лежит более
высокая термодинамическая стабильность разветвленных алкенов.
Правило Зайцева применимо к реакциям, протекающим и по
Е1 и по Е2-механизмам.
В несимметричных дигалогенэтанах направление
отщепления также соответствует правилу Зайцева. Однако в этом случае в основе
его лежит более высокая кислотность атомов водорода при наименее гидрированном,
углеродном атоме из-за наличия у него большего числа электрноакцепторных заместителей
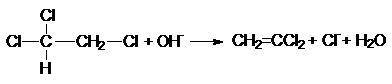
При разложении четвертичных аммониевых и сульфониевых
соединений направление отщепления противоположное и определяется правилом Гофмана,
согласно которому водород отщепляется от наиболее гидрированного атома углерода,
в результате чего образуется менее разветвленный алкен:
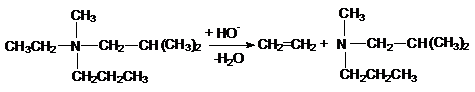
Такой порядок расщепления связан с близостью
механизмов этих реакций к механизму Е1св. В переходном состоянии
записанной реакции связь С-Н ослаблена сильнее, чем связь С-N, и промежуточный
карбанион (или близкое к нему по структуре переходное состояние) оказывается
менее дестабилизированным алкильными группами при образовании менее
разветвленного алкена.
Конкуренция
реакций нуклеофильного замещения и отщепления и роль изомеризации.
Если структура субстрата делает возможным его
расщепление, эта реакция всегда конкурирует с замещением. Очевидно, что состав
продуктов будет зависеть от соотношения скоростей этих конкурирующих реакций,
определяемых различными факторами. При конкуренции SN1– и Е1-реакций
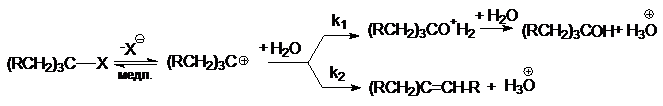
cостав продуктов не зависит от общей скорости и
определяется соотношением констант скоростей быстрых стадий k1/k2.
На состав продуктов не влияют ни способ образования карбкатиона, ни природа
отщепляемой группы Х, что может служить подтверждением SN1– и
Е1-механизма.
Удлинение и разветвление алкильной группы в исходном
соединении приводит к росту доли алкена в продуктах реакции при конкуренции SN1–
и Е1-механизмов. Это объясняется большей стабилизацией переходного состояния
образования более разветвленного алкена из-за его высокой термодинамической
стабильности. переход к более
сольватирующей среде способствует увеличению удельного веса продукта замещения,
так как переходное состояние быстрой стадии реакции SN1
характеризуется большей степенью локализации заряда
(RCH2)3Cd+…Od+H2
чем переходное состояние конкурирующей с ней реакции
отщепления
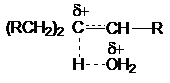
Благодаря этому первое сольватируется сильнее, что
приводит к большему понижению энергетического барьера реакции замещения.
При конкуренции реакций SN2 и Е2
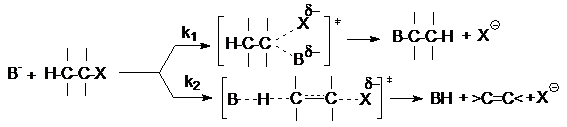
удлинение и разветвление алкильного радикала субстрата
приводит к увеличению доли продукта отщепления, как и в случае конкуренции SN1–E1.
В основе такого влияния лежат две причины: более высокая термодинамическая
стабильность разветвленных алкенов и возрастание стерических препятствий в
реакции SN2 с увеличением размера алкильной группы субстрата. В то
же время в реакции отщепления Е2 атака основания направлена на периферийный
атом водорода и практически не имеет стерических препятствий. Из приведенных
соображений следует, что при переводе механизма из SN1– Е1 в SN2– Е2 должен повышаться выход продукта
отщепления.
В реакции SN2 по сравнению с Е2 переходное
состояние характеризуется большей поляризацией отрицательного заряда, поскольку
в случае SN2 заряд распределен по трем атомам, а в случае Е2 – по
пяти. Поэтому при переходе к более сольватирующему растворителю энергия
переходного состояния SN2 будет понижаться в большей степени, чем
энергия переходного состояния Е2, что приводит к увеличению доли продукта замещения.
Наоборот, при использовании неполярных апротонных растворителей увеличивается
выход алкена.
Важным фактором регулирования состава продуктов
замещения-отщепления является температура. Как правило, энергия активации
реакции отщепления (Е2) выше, чем энергия активации замещения (SN2).
Поэтому увеличение температуры приводит к увеличению веса продукта отщепления.
Понижение температуры приводит к противоположному результату. В случае конкуренции
SN1– Е1 температура в меньшей степени влияет на состав продуктов,
так как различия в энергиях активации конкурирующих реакций из-за их экзотермичности
существенно ниже.
Большое значение в конкуренции реакций замещения и
отщепления имеет природа нуклеофильного реагента. Успех реакций SN2
определяется его сродством к углеродному атому, имеющему частичный положительный
заряд, т.е. нуклеофильностью. При Е2- и Е1св-отщеплении нуклеофил
атакует положительно заряженный атом водорода и его активность определяется его
основностью. Различие между нуклеофильностью и основностью часто является очень
большим и это во многом определяет направление реакции. Для таких сильных
оснований, как едкие щелочи и третичные амины наиболее характерны реакции
отщепления, что обусловливает их практическое применение для процессов
дегидрохлорирование. Наоборот, для относительно сильных нуклеофилов, но слабых оснований
(HS–, RS–, ArO–, NH3, Br–,
J–) преобладающим направлением является замещение. Этим пользуются
при промышленном гидролизе хлорпроизводных в спирты, когда применение едких щелочей
дает слишком большой выход алкенов. Замена щелочей на более слабое основание
(карбонат натрия, его буферные смеси со щелочью или бикарбонат натрия)
позволяет существенно повысить выход спиртов.
В случае конкуренции SN1– Е1, различия в нуклеофильности и основности
не играют такой существенной роли по двум причинам: во-первых, нуклеофильная
атака центрального атома углерода карбкатиона не встречает серьезных стерических
препятствий из-за плоского строения последнего; во-вторых, быстрая стадия
реакции SN1 в значительной степени определяются зарядовым контролем,
в котором роль основности существенно возрастают.
Таким образом, преимущественному замещению
способствует менее разветвленная структура алкильной группы в исходном соединении,
высокая нуклеофильность реагента по сравнению с его основностью, большая сольватирующая
способность среды и умеренная температура. Очевидно, что отщеплению благоприятствуют
противоположные условия.
Кроме рассмотренной конкуренции реакций реакции SN1
и Е1 могут сопровождаться побочными
реакциями изомеризации. причина
этих реакций состоит в том, что промежуточно образующийся карбкатион, прежде
чем прореагировать с соответствующими нуклеофилом, способен к внутримолекулярной
стабилизации за счет миграции к заряженному атому углерода отдельных атомов или
групп.
В результате образуется новый карбкатион (или
несколько карбкатионов), который при последующей реакции с нуклеофилом дает
смесь продуктов замещения и отщепления. Подобные перегруппировки могут
осуществляться следующими путями:
1. Миграция алкильных групп.
Мигрирующая группа R перемещается вместе со связующей
парой электронов и выполняет роль нуклеофила, внутримолекулярно атакующего реакционный
центр. Поэтому его способность к миграции зависит от нуклеофильности и
повышается при наличии в ней электронодонорных заместителей и с ростом их поляризуемости:
(CH3)3C-
> (CH3)2CH- > CH3CH2- > CH3-
2. Миграция атомов водорода (гидридный перенос).
Направление миграции атомов водорода определяется
относительной стабильностью ионов карбония, часто находящихся в равновесии друг
с другом. Поэтому преимущественно образуются третичные или вторичные продукты
замещения или более разветвленные алкены. Особенно часто такие перегруппировки
происходят при кислотнокаталитических превращениях спиртов. Так, при замещении
гидроксила в бутаноле-1 на галоген получается на первичный, а вторичный
бутилгалогенид:
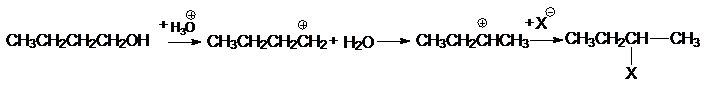
3. Перегруппировки, связанные с наличием в
интермедиатах нескольких реакционных центров.
Причина образования таких центров обусловлена
сопряжением вакантной орбитали карбкатиона с аллильной двойной связью или системой
сопряженных двойных связей. Так, при гидролизе кротилхлорида образующийся карбкатион
имеет два положительно заряженных реакционных центра. Поэтому при нуклеофильной
атаке такого карбкатиона образуются два продукта замещения
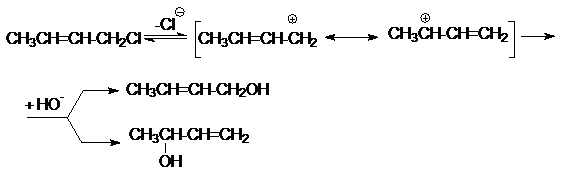
Для предотвращения всех типов изомеризации необходимо
перевести реакцию из области SN1–
Е1 в область SN2– Е2.
Этому способствуют выбор апротонных неполярных растворителей, применение электрофильных
катализаторов, снижение температуры и другие факторы, рассмотренные ранее.