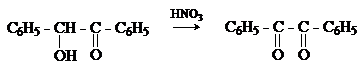Альдегиды и кетоны, их свойства и способы получения.
Карбонильные соединения – производные углеводородов, в
которых два атома водорода при одном и том же атоме
углерода замещены на атом кислорода. Результатом такого замещения является
наличие в структуре молекулы органического соединения карбонильной группы >С=О. Если одна из валентностей атома углерода этой
группы затрачена на образование химической связи с атомом водорода, то такая
группа называется альдегидной, а сами соединения, содержащие такую группу
называются альдегидами
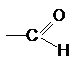
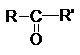
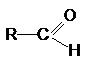
Если
обе валентности карбонильной группы затрачены на образование связей с
углеводородными радикалами, то такие соединения называются кетонами.
Если углеводородные радикалы в альдегидах и кетонах
являются алькильными, то такие альдегиды и кетоны
называются предельными или насыщенными.
Методы получения
альдегидов и кетонов.
1.
Окисление или
окислительное дегидрирование спиртов

2.
Озонолиз алкенов
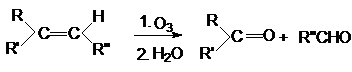
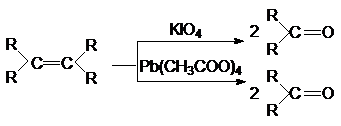
3.
Окисление алкенов
Pb(OOCCH3)4
и KIO4
4.
Каталитическое
окисление алкенов в присутствии комплексов палладия
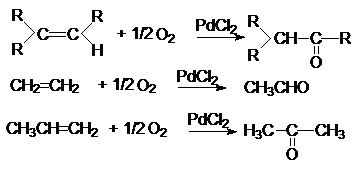
5.
Гидратация алкинов
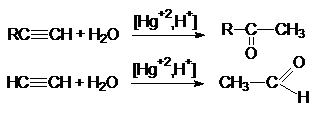
6.
Гидроборирование алкинов
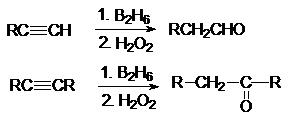
7.
Гидроформилирование (оксосинтез) алкенов
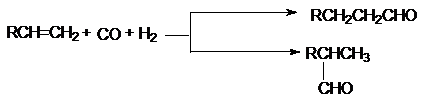
Условия: cat: HCo(CO)4,
150оС, 10-15 МПа.
8.
Гидролиз геминальных дигалогеналканов
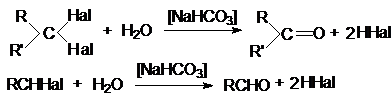
9.
Пинаколиновая перегруппировка
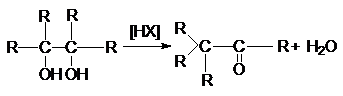
10.
Магний-органический синтез
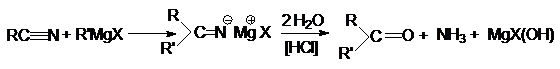
11.
Превращение
карбоновых кислот и их производных
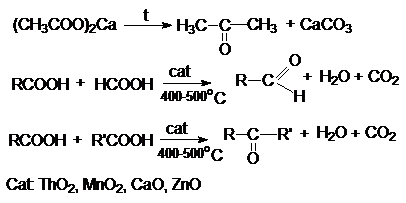
12.
Реакция Розенмунда
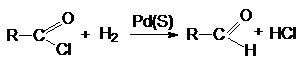
Насыщенные альдегиды и кетоны являются бесцветными
жидкостями со своеобразным запахом (формальдегид – газ с острым запахом).
Температуры кипения ниже, чем у соответствующих алканолов.
Карбонильная группа – сильно
полярная группа. В то же время она обладает значительной
поляризуемостью. Это можно проиллюстрировать данными
молекулярной рефракции связей. Так, для связей С–О R=1,5, а для связи С=О R=3,3 ... 3,5. Это означает, что на атомах карбонильной
группы не только имеются значительные эффективные заряды, но они еще
увеличиваются под действием внешних факторов (атакующих реагентов).
Положительный заряд на карбонильном углероде
обусловливает его восприимчивость к атаке нуклеофильными реагентами. В то же время карбонильный кислород обладает основностью и может благодаря этому протонироваться
под действием Бранстедовских кислот.
В УФ-спектрах
карбонильных соединений имеются два максимума поглощения: интенсивный в
диапазоне 150-200 нм (p – p*) и малоинтенсивный в
диапазоне 270-300 нм (n – p*).
Характерны четкие максимумы в ИК-спектрах
карбонильных соединений в области 1755 – 1695 см-1, которые связаны
с валентными колебаниями группы С=О, причем альдегиды
поглощают при более коротких волнах, чем кетоны.
ИК-спектры, а также спектры комбинационного рассеяния являются надежными
методами для обнаружения карбонильной группы.
В ПМР-спектрах альдегидный
протон имеет характерный сигнал = 8 ... 10,5 м.д., который обычно не
перекрывается с сигналами других протонов.
Химические свойства
альдегидов и кетонов.
Химическое поведение альдегидов и кетонов определяется
четырьмя факторами. Во-первых, положительный заряд карбонильного углерода
обусловливает его восприимчивость к нуклеофильной атаке. При этом основность карбонильного кислородного атома создает
возможность дополнительного увеличения положительного заряда на карбонильном
углероде за счет протонирования первого кислотами
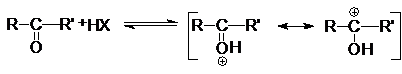
Благодаря этому карбонильный углерод может успешно
атаковаться даже слабыми нуклеофилами.
Подобная активация молекул карбонильных соединений реализуется в кислотном
катализе реакций нуклеофильного присоединения по карбонильной группе.
Во-вторых, слабость p-связи в
карбонильной группе обусловливает стехиометрический результат нуклеофильных
реакций этих соединений: уходящей группой при нуклеофильной атаке является
карбонильный атом кислорода, который сохраняет свою s-связь с карбонильным углеродным атомом.
В-третьих, электроноакцепторный характер карбонильной
группы за счет –I и –M–эффектов обусловливает кислотность водородных атомов при соседних с ней углеродных атомов, что проявляется в
способности последних отщеплять протон под действием сильных оснований

Можно видеть, что обоснованием кислотности водородных
атомов при a-углеродном атоме является
стабилизация образующегося карбаниона за счет –I и
–М–эффектов карбонильной группы.
Подвижность атомов водорода при a–углеродном атоме является предпосылкой для замещения
этих атомов другими заместителями или группами, что находит свое выражение в
реакциях a–галогенирования и
конденсации.
В-четвертых, промежуточная степень окисления
карбонильных групп (+1 в альдегидах и +2 в кетонах) обусловливает их
способность к реакциям окисления и восстановления.
Реакции нуклеофильного присоединения
по карбонильной группе.
Стехиометрия реакций нуклеофильного присоединения
реагента :NuH к карбонильному соединению представляется следующим
уравнением
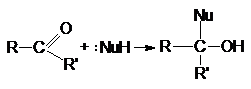
Как указывалось выше, атака нуклеофильного реагента на
карбонильный атом углерода приводит к разрыву p-связи
карбонильной группы. Образующийся при этом интермедиат претерпевает дальнейшие превращения, приводящие
к образованию продуктов нуклеофильного присоединения.
Реакции нуклеофильного присоединения по карбонильной
группе могут осуществляться при кислотном, основном катализе или некаталитически.
В некаталитической реакции реализуется механизм:
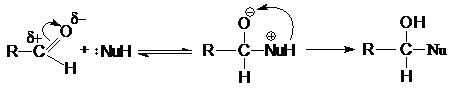
1.
Кислотно-каталитическая
реакция образования ацеталей и полуацеталей
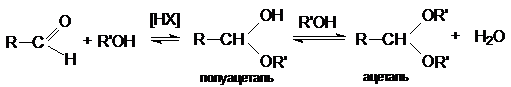
Механизм образования полуацеталей
согласуется с общим механизмом кислотно-каталитического нуклеофильного
присоединения по карбонильной группе
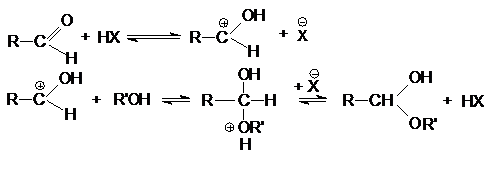
Вторичная реакция образования ацеталей
является типичной кислотно-каталитической реакцией образования простого эфира
2.
Образование
гидратов альдегидов.
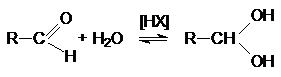
Механизм этой реакции подобен механизму образования полуацеталей. Положение равновесия зависит от структуры
альдегидов и условий реакции.
3.
Взаимодействие с
гидросульфитом (бисульфитом) натрия
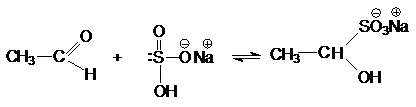
Катализатор в этой реакции не требуется, так как
гидросульфит – анион в ионной паре соли является достаточно эффективным нуклеофильным
агентом.
Продукты присоединения гидросульфита с карбонильным
соединением – хорошо кристаллизующиеся вещества, нерастворимые в водном
растворе гидросульфита натрия. Поэтому их используют для отделения карбонильных
соединений от веществ, с которыми гидросульфит натрия не вступает в реакции.
При нагревании с раствором соды или щелочей полученные аддукты
разлагаются с образованием сульфита натрия и регенерацией карбонильного соединения.
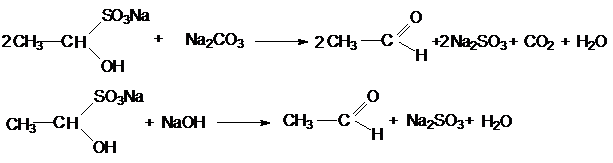
Следует отметить, что в реакцию с гидросульфитом
натрия вступают только альдегиды и метилкетоны.
4.
Замещение
кислорода карбонильной группы на галогены.
Галогенирующими агентами по отношению к альдегидам и кетонам выступают
галогениды фосфора, серы и др.
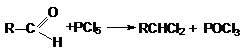
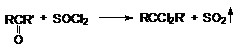
При взаимодействии с этими реагентами, которые
являются сильными электрофилами, происходит активация
карбонильного соединения, сопровождающаяся нуклеофильной атакой галогенид–аниона на карбонильный углерод
5.
Взаимодействие с
реактивами Гриньяра
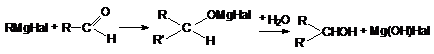
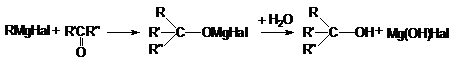
6.
Образование циангидринов

7.
Реакции
нуклеофильного присоединения азотистых оснований.
К этим реакциям относятся:
а)
образование иминов (азометинов)
– оснований Шиффа
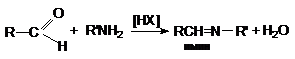
б)
образование оксимов
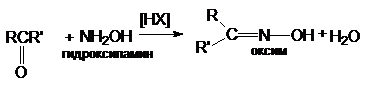
в) образование гидразонов

г) синтез семикарбазонов
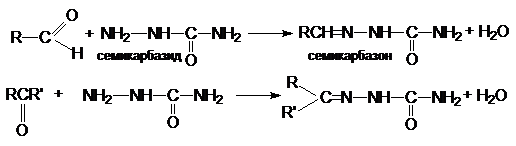
Реакционная способность карбонильных
соединений в реакциях
нуклеофильного присоединения.
Скорость-определяющей стадией реакций нуклеофильного присоединения по
карбонильной группе является бимолекулярная атака реакционного центра субстрата
нуклеофилом. Поэтому успех реакции определяется
величиной частичного положительного заряда на карбонильном углероде и его
пространственной доступностью. Поэтому реакционная способность карбонильных соединений
падает по мере увеличения длины и разветвленности алкильного радикала при карбонильном
углероде, а также при переходе от альдегидов к кетонам:
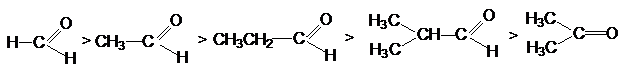
Можно видеть, что такое влияние связано с увеличением
в этом ряду положительного индуктивного эффекта алкильных групп и с возрастанием
пространственных препятствий для атаки на карбонильный углерод.
Кето–енольная таутомерия, катализируемая
основаниями и кислотами.
Кето-енольная таутомерия – форма изомерии, в которой изомеры (таутомеры) отличаются между собой расположением атомов водорода
и кратностью связей между другими атомами.
Для карбонильных соединений характерна таутомерия в связи
с кислотными свойствами атомов водорода при a-углеродном
атоме и основностью карбонильного кислорода. Исходная
карбонильная форма называется кето-формой,
а его таутомер – енольной
формой.
R-CH2CHO
« R-CH=CH-OH
кето-форма енольная форма
Взаимный переход таутомеров
также называется кето-енольной таутомерией. Эта
реакция катализируется основаниями и кислотами.
Основно-каталитическая реакция:
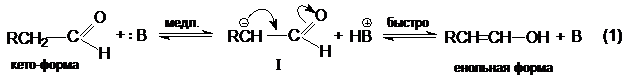
Кислотно-каталитическая реакция
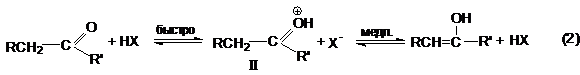
Присоединение протона к кислороду карбонильной группы
резко облегчает отрыв протона от a-углеродного
атома вследствие большей электроноакцепторной способности положительно
заряженного кислорода.
Кето-енольная таутомерия играет важную роль во многих превращениях
альдегидов и кетонов, так как енолы, а также интермедиаты I и II являются важнейшими интермедиатами
в реакциях альдегидов и кетонов.
Рассмотрим некоторые из этих реакций:
Результатом галогенирования является замещение
водорода при a-углеродном атоме по
отношению к карбонильной группе
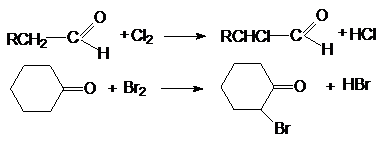
2. Альдольная конденсация
Реакции альдольной
конденсации могут быть представлены стехиометрическим уравнением
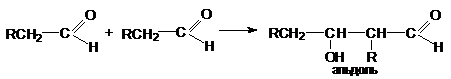
Альдольная конденсация
кетонов.
Кетоны значительно труднее вступают в реакции альдольной автоконденсации по
сравнению с альдегидами. Структурные условия здесь неблагоприятны как с точки
зрения скорости, так и сточки зрения равновесия. Однако если реакцию проводить
при кислотном катализе, то образующийся продукт конденсации будет быстро дегидратироваться, превращаясь в мезитилоксид.
Эта последняя стадия смещает равновесие в сторону полного превращения кетона
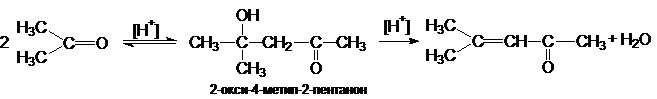
Обмен водорода при а-углеродных
атомах карбонильных соединениях на дейтерий под действием тяжелой воды может
быть представлен стехиометрическим уравнением
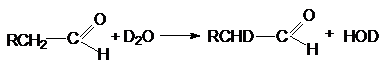
Эта реакция катализируется кислотами и основаниями.
Кислотно-каталитическая реакция предполагает ключевую роль енольной
формы карбонильного соединения
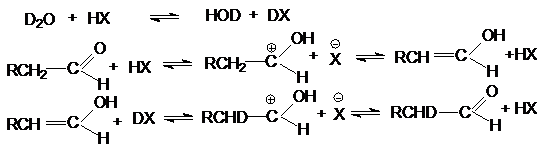
Первичная реакция изотопного обмена может
сопровождаться последующим дейтерированием, если при a-углеродном атоме остаются незамещенные атомы водорода.
Катализ изотопного обмена основаниями протекает через
следующие стадии

4. Рацемизация оптически активных кетонов
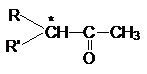
Рацемизация таких оптически активных соединений
катализируется кислотами и основаниями. Основно-каталитическая рацемизация
может быть легко обоснована образованием карбаниона
при отрыве протона основанием от хлорального углеродного
атома.
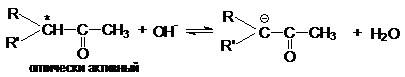
Поскольку оптически активный карбанион
имеет плоское строение последующая атака воды при ее протолитическом
взаимодействии с a-углеродным
атомом равновероятна с противоположных сторон и поэтому дает рацемическую
смесь.
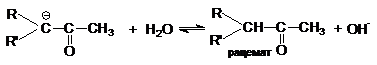
Кислотно-каталитическая рацемизация приводит к образованию енольной формы кетона, в которой хиральность
атома углерода утрачивается, поэтому обратная реакция образования кето-формы дает рацемическую
смесь.
Промежуточная степень окисления карбонильных
соединений обусловливает их способность к реакциям окисления и восстановления.
В качестве восстановителей в реакциях восстановления используют молекулярный
водород и гидридные комплексы алюминия и бора.
1.
Восстановление
карбонильных соединений до спиртов.
а)
восстановление молекулярным водородом
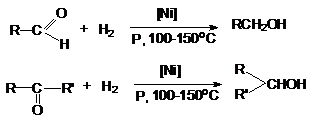
В качестве катализаторов этих реакций используют Ni, Pd, Pt,CuCrO2.
б) восстановление гидридными
комплексами металлов
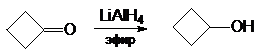
2.
Восстановление
карбонильных соединений до углеводородов.
а) восстановление по Клемменсену.
Если карбонильное соединение устойчиво к действию
кислот, то используют этот тип восстановления
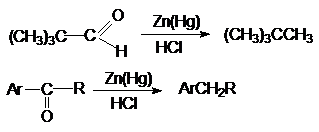
б) восстановление по Кижнеру-Вольфу
Этот вид восстановления используется в тех случаях,
когда объект восстановления устойчив к основаниям
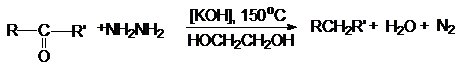
Реакции окисления карбонильных
соединений.
Альдегиды легко окисляются органическими окислителями
(KMnO4 в нейтральной и кислой среде , K2Cr2O7 в кислой
среде) до карбоновых кислот

В промышленном масштабе окисление осуществляется
кислородом воздуха
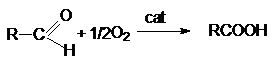
при катализе солями Mn4+, Co3+, Ni3+ и др.
Кетоны к действию окислителей весьма устойчивы и
окисляются лишь сильными окислителями при нагревании. Реакция имеет деструктивный
характер: при окислении происходит разрыв С-С связи по
обе стороны карбонильной группы
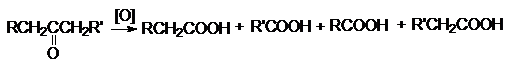
В этих реакциях используют те же окислители, что и при
окислении альдегидов: неорганические окислители в препаративном
органическом синтезе и кислород в промышленности. Окисление по Байеру-Виллигеру предполагает использование в качестве
окислителей органических перкислот
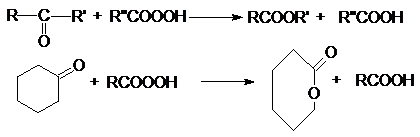
Реакции окислительно-восстановительного диспропорционирования.
В этих реакциях карбонильная группа проявляет двойственную функцию –
окислителя и восстановителя.
1.
Реакция Канницциаро
Альдегиды, которые не содержат
водородных атомов при карбонильной группе реагируют в концентрированном
водном растворе щелочи, давая 1 моль спирта и 1 моль карбоновой кислоты (в виде
соли)
2RCHO + NaOH RCH2OH
+ RCOONa
2.
Реакция Тищенко.
![]()
3.
Аминометилирование по Манниху
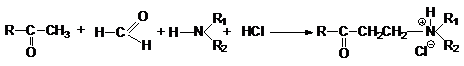
Обычно реакцию проводят в присутствии кислоты и поэтому продукт получают
в виде соответствующей соли. Механизм заключается в последовательности стадий:
Дикетоны. a, b - Непредельные альдегиды и кетоны. Ароматические альдегиды
и кетоны.
1,3–дикарбонильные соединения обладают высокоподвижным атомом водорода
при центральном углероде. Как следствие, эти соединения легко подвергаются енолизации.
Енольная и кето-формы,
а также енолят-анион являются ключевыми частицами,
ответственными за реакции этих соединений.
К наиболее важным химическим свойствам
1,3-дикарбонильных соединений относятся:
Кислотность и енолизация.
1,3 – дикетоны проявляют
кислотные свойства, причем за кислотность отвечают две формы – кето– и енольная.
Равновесие между этими таутомерными формами
устанавливается через промежуточное образование енолят
– аниона.
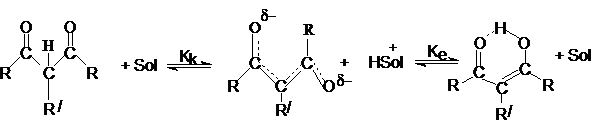
Дикарбонильняя форма является С-Н-кислотой, а енольная
– ОН–кислотой. Каждая из форм имеет свою константу
кислотности: Кк и Ке.
Чтобы оценить Кк и Ке,
необходимо знать соотношения таутомерных форм, т.е константу таутомерного равновесия, Кт.
Кт = [E]/[K], где [E] и [K] –
концентрации таутомерных форм.
Можно показать, что Кт = Кк / Ке (1)
Из соотношения (1) видно, что доминирующей таутомерной формой является та, которая имеет меньшую
кислотность. Так, для ацетилацетона рКк =
8,9, а рКе = 8,2, откуда следует, что Кт
= 0,2, что соответствует содержанию енола 16,5%. На
соотношение констант кислотности таутомеров влияют растворитель
и структура соединения.
1.
Влияние
растворителя.
В общем случае растворитель по-разному влияет на
кислотность таутмерных форм, поэтому при переходе от
одного растворителя к другому меняется соотношение таутомерных
форм. Например, в водном растворе для ацетилацетона Кт = 0,2, а в
растворе метанола Кт = 2,6 (72% енола).
Такой результат можно обосновать с точки зрения различия в кислотностях воды и
метанола. Так, вода, обладающая более высокой кислотностью, чем метанол, образует
более прочные водородные связи с кето–формой ацетилацетона. В то же время кислотность
растворителя не играет такой существенной роли в сольватации циклической енольной формы, так как в последней потенциал карбонильного
атома кислорода использован для образования внутримолекулярной водородной
связи. Как следствие, в воде более выгодной становится существенно сольватированная кето–форма.
2. Влияние структуры дикетона
Структура дикетонов в общем
случае по–разному влияет на относительную устойчивость кето- и енольной форм. Так,
шестичленные циклические транс– фиксированные b-дикетоны сильно енолизированы, так как у енольной
формы идеально соблюдается условие компланарности для
сопряжения, а, следовательно, стабилизации.
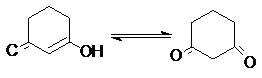
100% енола
В то же время такие условия отсутствуют у бициклических 1,3–дикетонов с центральным
атомом углерода в голове моста.
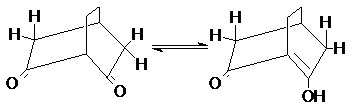
Следствием
кислотности 1,3–дикетонов является образование ими
солей при взаимодействии с активными металлами или сильными основаниями.

Особенно
характерны соли тяжелых металлов, которые в растворах практически не диссоциируют. Благодаря этому в качестве реагентов для
синтеза таких солей можно использовать соли тяжелых металлов – сильные электролиты.

Взаимодествие с электрофильными
реагентами.
Анион, ионная пара соли или енольная
форма восприимчивы к действию электрофильных агентов.
Примером могут быть реакции алкилирования или ацилирования. Как енол, так и
анион являются амбидентными частицами. Местом электрофильной атаки в них могут быть кислородные атомы или
углеродный атом между карбонильными группами (активная метиленовая группа).
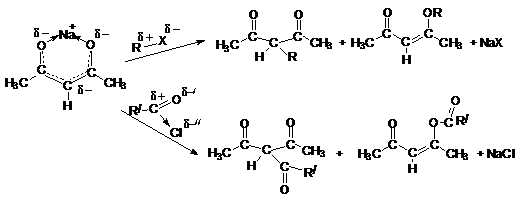
В общем случае получается смесь продуктов С- и О-алкилирования или ацилирования. В случае 1,3–дикарбонильных
соединений, образующих цис–енольную форму, преимущественно образуются С-производные,
так как атомы кислорода значительно блокированны ионом
металла. Если обьектом электрофильного
замещения являются транс- фиксированные 1,3–дикетоны, то преимущественно могут получаться продукты О-
замещения.
Взаимодействие с другими электрофильными
реагентами (нитрование, сульфирование, галогенирование, нитрозирование,
азосочетание) может быть изображено общей схемой.
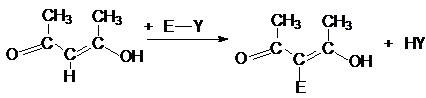
где Е – NO, NO2,
ArN2, SO3H, Cl, Br, I.
Взаимодействие с нуклеофильными реагентами.
Енольная или дикарбонильная форма
1,3–дикетонов могут быть объектами нуклеофильной
атаки, причем атакующим реакционным центром в этих реакциях выступает
карбонильный углерод. Если в реакциях
участвуют слабые нуклеофилы (вода, спирты, тиолы), то для успешной реализации реакции используют кислотный
катализ.
Примером нуклеофильной атаки является взаимодействие с
аммиаком и аминами. Считается, что активной формой, реагирующей с RNH2,
является дикарбонильная форма.
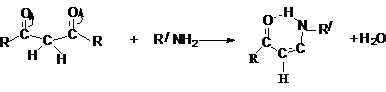
Результатом этих реакций является образование енаминов, стабилизированных за счет образования прочной
внутримолекулярной водородной связи.
В случае реакции с гидроксиламином и гидразинами образуются гетероциклические соединения. Подробное описание этих реакций будет дано в материале по гетероциклическим соединениям.
Примером другой реакции нуклеофильного замещения
является щелочной гидролиз 1,3 – дикетонов.
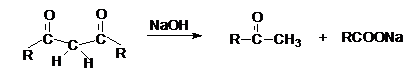
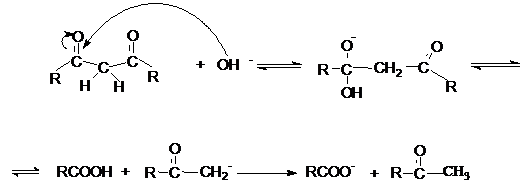
Реакция может быть представлена механизмами.
a,b - Непредельные альдегиды и кетоны.
Соединения этого ряда могут быть представлены
структурой
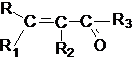
где R ,R1 ,R2 ,R3 -
представляют собой алкильные группы или атом водорода.
1. Дегидратация
альдолей, кротоновая
конденсация
Альдоли –
продукты альдольной конденсации, легко отщепляют
воду, особенно при кислотном катализе.
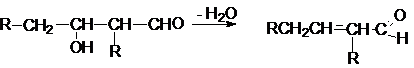
В результате дегидратации альдоля
получается кротоновый альдегид, поэтому часто этот
процесс называют кротонизацией.
В ряде реакций карбонильных соединений, например, аренкарбальдегидов с кетонами, продукт альдольного
присоединения выделить не удается и сразу получается ненасыщенные соединения.
Такие реакции называют кротоновой конденсацией.
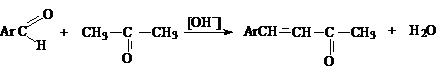
1. Реакции
окисления алкенов и алкинов
Использование некоторых окислителей, например, CrO3 при
низких температурах, позволяет селективно окислять спиртовые группы до альдегидных и кетонных:
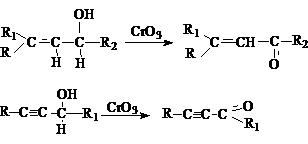
Другой вариант подобного синтеза – каталитическое
окисление кислородом
![]()
Структурные особенности a, b -
ненасыщенных карбонильных соединений.
Соединения характеризуются полярной сопряженной
системой. В виду значительного индуктивного (- I) эффекта и большого изомерного (- М)
эффекта карбонильной группы двойная углерод – углеродная связь сильно поляризована.
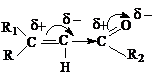
Дипольный момент таких
соединений больше, чем для насыщенных карбонильных соединений. a,b-Ненасыщенные карбонильные соединения в молекуле имеют два электрофильных реакционных центра.
a, b-ненасыщенные карбонильные соединения могут вступать в
обычные реакции присоединения и конденсации по карбонильной группе, такие как
образование циангидридов и гидразонов
и присоединение металлоорганических соединений. Эти реакции могут осложняться,
а иногда перекрываться 1,4–присоединением.
1.
Циангидринирование
Реакция циановодорода с альдегидами протекает
как 1,2–присоединение, так как этот путь более выгоден с точки зрения скорости
и с точки зрения равновесия, чем 1,4–присоединение.
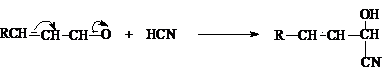
Образование циангидринов
кетонов менее выгодно с точки зрения скорости и равновесия и в результате
1,4–присоединение приводит к b-цианокетону
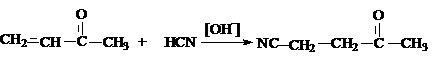
Механизм

2.
Присоединение
воды и галогеноводородов к a, b - ненасыщенным альдегидам и
кетонам

В кислой среде α, β–ненасыщенные альдегиды и
кетоны присоединяют воду по связи С=С против правила Марковникова. Однако первичным взаимодействием в этой
реакции является 1,4–присоединение.
Аналогично идет присоединение кислот НХ.
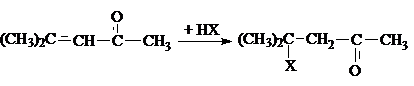
3.
Реакции
нуклеофильного присоединения
а) взаимодействие с Mg–органическими соединениями
Альдегиды и кетоны ведут себя по разному
с Mg–органическими соединениями.
Альдегиды присоединяют их по >C=O
связи, как и насыщенные альдегиды.
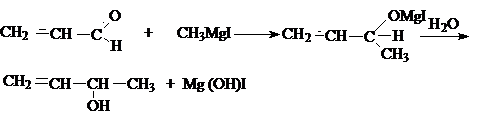
Кетоны реагируют по типу 1,4-присоединения.
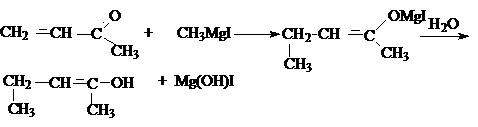
б) присоединение аммиака и аминов идет по типу
1,4–присоединения, которое в силу неустойчивости интермедиатов превращается в продукт 3,4–присоединения.
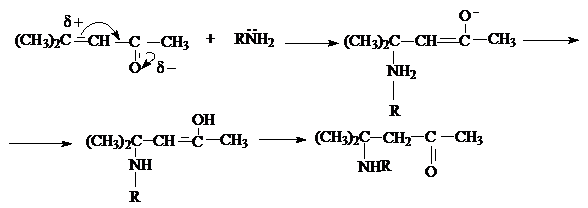
в) присоединение триалкилборатов.
Эта реакция приводит к образованию b-алкил- замещенных насыщенных альдегидов или кетонов.
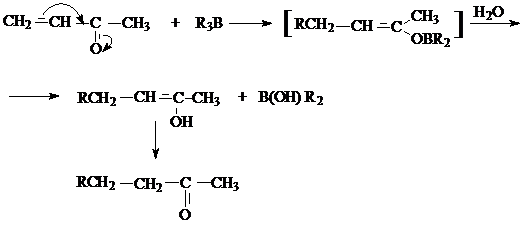
г) присоединение литийорганических соединений.
В отличие от триалкилборатов
литийорганические соединения реагируют по типу присоединения по карбонильной
группе:
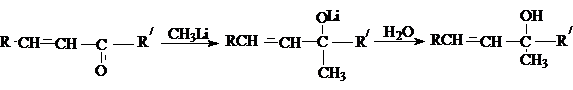
д) присоединение
медь-органических соединений
Медь–oрганические
соединения присоединяются по типу 1,4–присоединения.
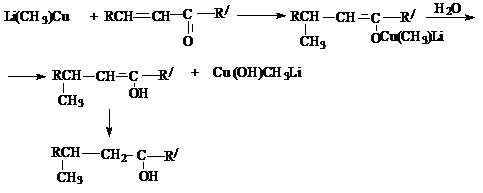
е) реакции Михаэля – сопряженное присоединение нуклеофилов к a,b- ненасыщенным системам.
Механизм:
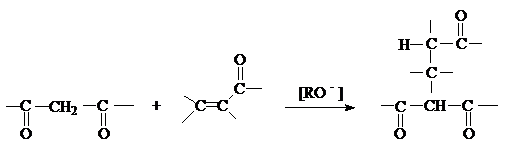
Восстановление a,b-ненасыщенных соединений осуществляется в разных
направлениях в зависимости от типа восстановителя.
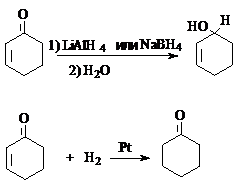
Ароматические альдегиды и кетоны.
Различают следующие типы
карбонильных соединений аренов.
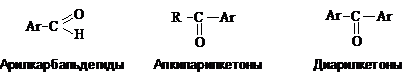
При составлении названий ароматических карбонильных
соединений используют заместительную номенклатуру IUPAC, рациональную номенклатуру и тривиальные названия.
1)
Реакции окисления
Аренкарбальдегиды получают окислением метиларенов
и бензиловых спиртов.
ArCH3 ArCHO
![]()
Окислители: CrO3 + (CH3CO)2O; CrO2Cl2
При окисление других алкиларенов образуются алкиларил-
кетоны.
ArCH2R ArC(O)R
Окислители: О2 в
присутствии катализаторов – солей кобальта и марганца.
Окисление диарилметанов дает
диарилкетоны.
ArCH2Ar ArC(O)Ar
Окислители: KMnO4; CrO3.
2)
Гидролиз дигалогеналкиларенов
ArCCl2R ArC(O)R
(R=H, Alk, Ar)
3)
Ацилирование аренов
ArH ArC(O)R
4)
Формилирование аренов
Для формилирования (введения
группы СНО) необходим хлорангидрид муравьиной
кислоты, который крайне нестабилен и разлагается на СО и НCl, поэтому используют другие формилирующие
реагенты
Реакция Гаттермана-Коха. Формилирование по Гаттерману-Коху осуществляется под действием оксида
углерода (II) и хлористого водорода в присутствии катализатора Фриделя-Крафтса - хлорида алюминия, промотированного
хлоридом меди (I).
![]()
Роль однохлористой меди в этой реакции неясна: возможно она связывает СО в комплекс, что способствует
образованию крайне нестабильного хлористого формила HCOCl из CO и HCl. Таким путем
удается ввести альдегидную группу в алкилбензолы, арилгалогениды, полициклические углеводороды, причем формильная группа вводится селективно в пара-положение.
Реакция не применима для формилирования фенолов, их
эфиров и ариламинов.
Реакция Гаттермана. В
качестве формилирующего агента используется смесь
безводного HCN и газообразного хлористого водорода в присутствии кислоты
Льюиса.
ArH + HCN + HCl ArCH=NHCl- ArC(O)H
Электрофильным реагентом является, вероятно, комплекс катализатора с
формимидхлоридом.
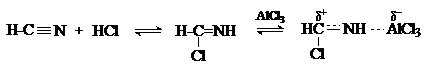
Образующийся в синтезе гидрохлорид альдимина
ArCH=NHCl гидролизуют
до альдегида.
Для того, чтобы избежать
применения ядовитой синильной кислоты, Р.Адамс модифицировал реакцию, заменив
синильную кислоту цианидом цинка (реакция
Гаттермана-Адамса). Это позволило из цианида
цинка и HCl получать непосредственно в реакционной
смеси HCN и безводный хлористый цинк, играющий роль слабой кислоты Льюиса. Этот
метод дает хорошие результаты при формилировании
фенолов и простых эфиров фенолов.
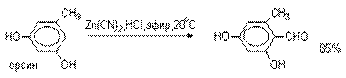

Реакции Гаттермана и Гаттермана-Адамса
используют также для формилирования алкилбензолов и конденсированных ароматических
углеводородов. Метод не применим для аренов,
содержащих дезактивирующие заместители, и ароматических аминов.
Реакция Вильсмейера-Хаака. В качестве формилирующего
реагента используют диметилформамид (ДМФА) в
присутствии POCl3 как кислоты Льюиса.
ArH + HC(O)N(CH3)2 … ArC(O)H
+ NH(CH3)2
Электрофильным агентом в реакции Вильсмейера-Хаака является иминиевая соль, которая образуется при взаимодействии ДМФА и хлорокиси фосфора.
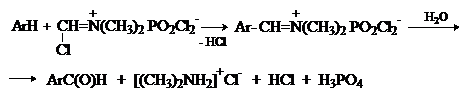
Реакция Вильсмейера-Хаака
чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие
выходы ароматических альдегидов, содержащих NR2-,OR- или OH-группы.
Она оказывается практически ценной при формилировании
конденсированных ароматических углеводородов - антрацена, азулена,
пирена и др., а также разнообразных гетероциклических
соединений ряда фурана, тиофена, пиррола, индола.
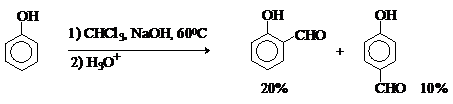
Реакция Реймера-Тимана. Используется для формилирования
фенолов.
5)
Восстановление хлорангидридов
аренкарбоновых кислот (реакция Розенмунда)
ArC(O)Cl + H2 ARC(O)H + HCl
Реакции ароматических карбонильных соединений подобны превращениям их
алифатических аналогов. Однако необходимо отметить следующие особенности:
-
карбонильные
соединения аренов обладают более низкой реакционной
способностью по отношению к нуклеофильным реагентам;
-
аренкарбальдегиды и диарилкетоны не способны
к енолизации;
-
возможны реакции электрофильного замещения в ароматическом кольце.
а)
Бензоиновая конденсация
При нагревании в присутствии цианидов аренкарбальдегиды образуют a-гидроксикетоны (бензоины).
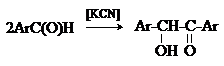
Механизм реакции:
Ключевая стадия процесса – образование карбаниона за счет миграции протона от атома углерода к атому кислорода, которая становится возможной благодаря электроноакцепторному действию цианогруппы.
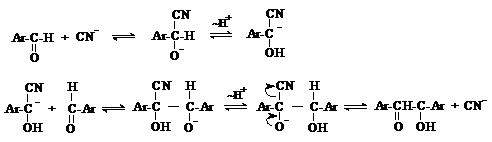
б) Реакция Канницаро
Аренкарбальдегиды, как альдегиды, не содержащие a-водородных атомов, вступают в реакцию Канницаро.
2ArC(O)H + NaOH ® ARCOONa
+ ARCH2OH
в) Реакция Перкина
Аренкарбальдегиды конденсируются с ангидридами карбоновых кислот в присутствии оснований
(ацетатов и карбонатов щелочных металлов, пиридина). При этом альдегид выполняет роль карбонильной, а ангидрид – метиленовой компоненты.
ArC(O)H + (CH3CO)2O Ar-CH=CH-COOH + CH3COOH + H2O
Механизм реакции:

г) Автоокисление
Аренкарбальдегиды очень легко окисляются кислородом воздуха на свету. Процесс протекает по свободнорадикальному механизму через промежуточное образование стабильных ароильных радикалов.
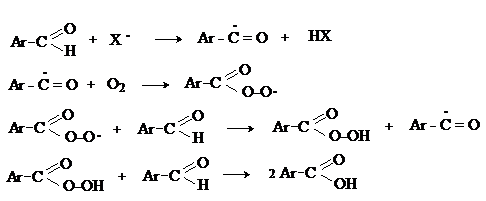
д)
Хлорирование
Аренкарбальдегиды легко хлорируются по свободнорадикальному механизму с образованием хлорангидридов.
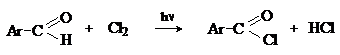
Механизм:
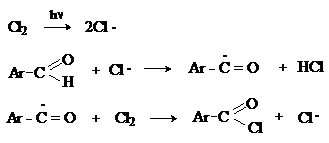
Важнейшие
представители.
Бензальдегид – бесцветная жидкость с запахом горького миндаля.
Получают из толуола прямым окислением или хлорированием до бензальхлорида
с последующим гидролизом. Как альдегид, не содержащий a-водородных
атомов, бензальдегид выступает в реакциях конденсации
в качестве метиленовой компоненты. При конденсации его с ацетальдегидом
образуется коричный альдегид:
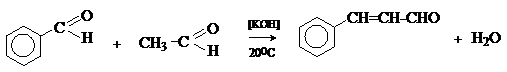
а при конденсации с с ацетофеноном – халкон:
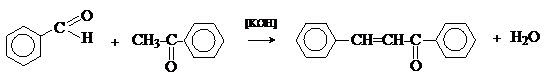
Коричный альдегид обладает свойствами a,b-непредельных карбонильных соединений. Используется в парфюмерии и
пищевой промышленности.
Бензофенон – бесцветное кристаллическое вещество. В
промышленности его получают окислением дифенилметана.
Для бензофенона характерно образование стабильного
анион-радикала синего цвета при взаимодействии с металлическим натрием.
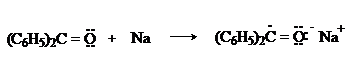
Такие анион-радикальные
соли называют металлкетилами.
Их относительная устойчивость связана со стабилизирующим влиянием на свободнорадикальный центр двух бензольных
ядер и отрицательно заряженного атома кислорода.
Дибензоил – желтое кристаллическое вещество. Образуется при
окислении бензоина.
При нагревании со спиртовым раствором щелочи дибензоил претерпевает перегруппировку с образованием бензиловой кислоты (бензиловая перегруппировка).
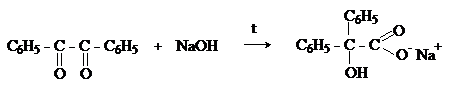
Механизм: