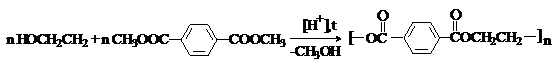Карбоновые
кислоты. Физико-химические свойства кислот: ассоциация и диссоциация,
химические свойства карбоновых кислот. Производные карбоновых кислот,
ангидриды, сложные эфиры, амиды, нитрилы.
Карбоновые кислоты являются производными углеводородов, в которых один
или несколько атомов водорода замещены на карбоксильную группу.
Карбоновые кислоты можно подразделить на две основные
группы:
1.
Монокарбоновые
кислоты (насыщенные, ненасыщенные,
аренкарбоновые кислоты).
2.
Ди- и
поликарбоновые кислоты (ненасыщенные, насыщенные, арендикарбоновые и
поликарбоновые кислоты).
Классификация,
изомерия и номенклатура.
Монокарбоновые кислоты подразделяют в зависимости от природы углеводородного
остатка.
a) Насыщенные монокарбоновые кислоты (производные алканов
и циклоалканов):
CnH2n+1COOH ,
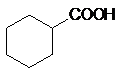
б) Ненасыщенные монокарбоновые кислоты (производные алкинов, алкенов, алкадиенов
и других ненасыщенных углеводородов,
CnH2n-1COOH
, CnH2n-3COOH и др.
в) Аренмонокарбоновые кислоты
ArCOOH, ArCH2COOH,
ArCH=CHCOOH
Методы
получения насыщенных монокарбоновых кислот.
1.
Окисление первичных спиртов и альдегидов.
В промышленном масштабе окисление ведут
кислородом воздуха при катализе солями
марганца или кобальта
RCH2OH +
O2 ![]() RCOOH + H2O
RCOOH + H2O
RCHO + 1/2O2 ![]() RCOOH
RCOOH
В лаборатории окисление осуществляют
неорганическими окислителями: KmnO4 в кислой или нейтральной среде, K2Cr2O7 в кислой среде.
2.
Окисление неразветвленных алкенов
RCH=CHR' RCOOH + R'COOH
Окислителями являются K2Cr2O7 в кислой среде или KmnO4 в кислой среде.
3. Окисление алкинов.
В качестве окислителей используют те же реагенты, что и при окислении алкинов.
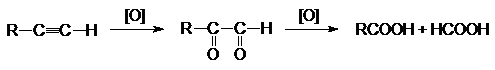
4. Окисление алканов (промышленный метод)
RCH2CH2R'
+ 5/2O2 ® RCOOH + R'COOH + H2O
Окисление осуществляют при катализе реакции солями
кобальта или марганца.
5.
Гидролитические методы.
а) Кислотный гидролиз нитрилов
RCºN + 2H2O
+ HX ® RCOOH + NH4X
б) Основной гидролиз нитрилов
RCºN + H2O
+ NaOH ® RCOONa + NH3
в) Кислотный гидролиз амидов кислот
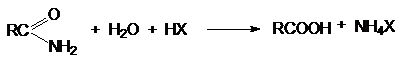
г) Основной гидролиз амидов кислот
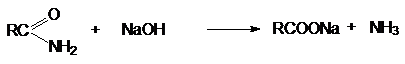
![]() д) Кислотный гидролиз сложных эфиров
д) Кислотный гидролиз сложных эфиров
RCOOR' + H2O
RCOOH + R'OH
е) Основной гидролиз сложных эфиров
RCOOR' + NaOH ® RCOONa + R'OH
ж) Гидролиз ангидридов карбоновых кислот
(RCO)2O
+ H2O ® 2RCOOH
з) Гидролиз галогенангидридов карбоновых кислот
RCOHal + H2O
® RCOOH + HHal
и) Гидролиз соединений, содержащих трихлорметильную
группу
RCCl3 +
2H2O ® RCOOH + 3HCl
Гидролиз осуществляет либо основанием, либо водой при
катализе апротонными кислотами, например, FeCl3.
6. Металлоорганический
синтез.
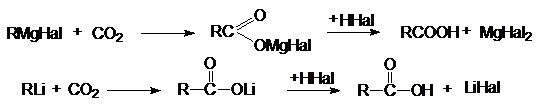
7. Реакции
карбонилирования
ROH + CO RCOOH
8.
Гидрокарбрксилироание галогеналканов и алкенов.
RHal + CO + H2O RCOOH + HHal
CH2=CH2
+CO+ H2O CH3CH2COOH
9. Синтез
Арндта-Эйстерта
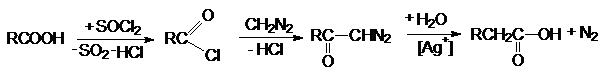
диазокетон
эта реакция представляет собой удобный способ
превращения карбоновой кислоты RCOOH в ее
ближайший гомолог RCH2COOH.
Технически важную муравьиную и уксусную кислоты
получают следующими способами.
Муравьиная
кислота и ее эфиры:
NaOH + CO HCOONa HCOOH
CO + ROH HCOOR
Уксусная кислота
CH3OH +
CO CH3COOH
CH3CHO
+1/2O2 ![]() CH3COOH
CH3COOH
Физические
свойства и строение монокарбоновых кислот.
Насыщенные монокарбоновые кислоты представляют собой бесцветные жидкие
или кристаллические вещества с острым своеобразным запахом, высшие карбоновые
кислоты (С15 – С18) имеют слабый запах стеарина. Они
имеют весьма высокие температуры кипения, что свидетельствует о значительной
межмолекулярной ассоциации следствие образования межмолекулярных водородных
связей, причем образуются как циклические димеры, так и линейные олигомеры.
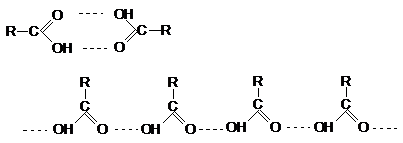
Электронографическое изучение карбоновых кислот показало, что в их
молекуле имеются карбонильная и гидроксильная группа, при этом связь С = О
длиннее, чем в кетонах, а связь С – О короче, чем в спиртах. Это свидетельствует
о сопряжении неподеленной пары кислорода гидроксильной группы и орбиталей карбонильной
группы
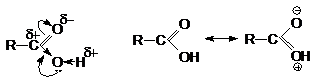
Проявляя +М – эффект, атом кислорода гидроксильной группы увеличивает полярность
связей ОН, но в то же время в некоторой степени уменьшает положительный заряд
на углеродном атоме по сравнению альдегидами и кетонами. Одновременно действует
электроноакцепторный индуктивный эффект (-I) кислородных атомов.
Таким образом в карбоксильной группе имеется сильно поляризованный положительно
атом водорода гидроксильной группы и углеродный атом которые являются
нуклеофильными центрами. В то же время кислородный атом имеет нуклеофильный
характер.
Химические
свойства карбоновых кислот.
Большинство реакций карбоновых кислот может быть отнесено к одному из четырех
основных типов:
1. Реакции, сопровождающиеся разрывом О-Н-связей,
например, кислотная диссоциация.
2. Реакции по карбонильному углероду, которые имеют
нуклеофильный характер.
3. Реакции декарбоксилирования
4. Реакции по a-углеродному атому алкильной группы.
Кислотность карбоновых кислот.
По сравнению со спиртами карбоновые кислоты обладают более высокой кислотностью.
При этом в растворе реализуется равновесие.
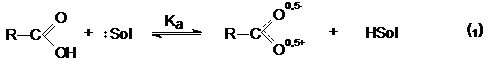
Способность к отдаче к протонам обусловлена двумя факторами:
поляризацией связей О-Н в исходном состоянии и стабилизацией карбоксилат аниона
из-за деколализации отрицательного заряда в нем посредством резонанса.
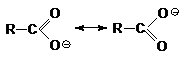
Кислотность карбоновых кислот характеризуется константой кислотности Ка
или показателем рКа. Их значения обычно составляет приблизительно 10-5,
что свидетельствует о слабости этих кислот. Присутствие в структуре радикала
электроноакцепторных заместителей будет стабилизировать карбоксилат анион, что
приведет к увеличению кислотности соответствующей ему кислоты. Например: рКа
монохлоруксусной кислоты составляет 2,9 а уксусной – 4,8. По мере удаления электроноакцепторного
заместителя от карбоксильной группы кислотность снижается, что связано с
быстрым затуханием индуктивного эфекта: так рКа b-протоновой кислоты составляет ~4,0. Наоборот увеличение разветвления алкильного радикала приводит к
снижению кислотности, так как при этом возрастает индуктивный эффект алкильной
группы. На основе этих данных можно представить следующие ряды кислотности
карбоновых кислот.
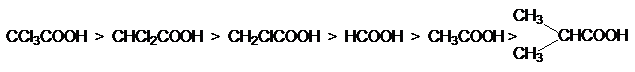
![]()
Кислотность карбоновых кислот проявляется в их взаимодействии
с активными металлами и основаниями.
2RCOOH + 2Na ® 2RCOONa + H2
RCOOH + NaOH ® RCOONa + H2O
2RCOOH + Na2CO3
® 2RCOONa + H2O
+ CO2
В кислой среде (pH<3) диссоциация карбоновых кислот
практически не происходит, так как равновесие (1) существенно смещено в левую
сторону из-за избытка ионов HSolÅ. В
то же время в кислой среде осуществляется протонирование карбоновых кислот по
основному карбонильному кислороду.
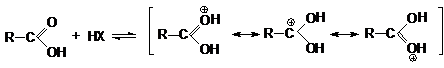
В протонированной форме оба кислородных атомов становятся одинаковыми:
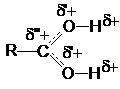
Несмотря на малую концентрацию протонированной формы
присутствие небольших количеств сильных кислот исключительным образом влияет на
реакционную способность карбоновых кислот: повышение положительного заряда на
карбонильном углероде делает его восприимчивым к атаке нуклеофильными реагентами
и открывает возможность для протекания многих реакций нуклеофильного замещения
карбоновых кислот.
Реакции нуклеофильного замещения.
1. Реакции этерификации
RCOOH + R'OH ![]() RCOOR' + H2O
RCOOR' + H2O
В механизме этой реакции ключевую роль играет
протонирование карбоновой кислоты кислотой-катализатором.
Реакция этерификации может осуществляться некаталитически,
однако для этого требуется жесткие условия процесса.
2.
Реакции
с N-нуклеофилами
(аммиаком, аминами, гидразином и др.).
N-нуклеофилы при взаимодействии с карбоновыми кислотами как
правило образуют аммониевые соли (карбоксилаты) и только при повышенных температурах
происходит присоединение N-нуклеофила к карбонильному углероду.
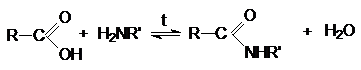
Механизм реакции согласуется с общепринятыми
представлениями о механизме нуклеофильного присоединения азотистых оснований по
карбонильной группе:
3. Взаимодействие с галогенангидридами
хлорорганических кислот (PCl3, PCl5, PBr3,
SOCl2).
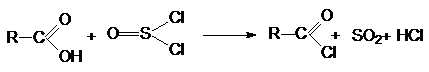
Механизм реакции связан с концертной атакой электрофильного
и нуклеофильного центров карбоновой кислоты молекулой галогенангидрида:
1.
Синтез Кольбе (см. подробно методы
получения алканов).
2RCOONa
+ 2H2O R-R +
2CO2 +2NaOH +H2
2.
Пиролиз карбоновых кислот.
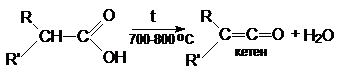
3.
Пиролитическая кетонизация солей
карбоновых кислот
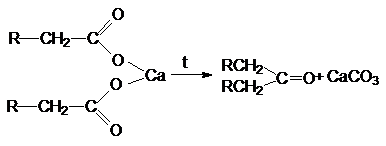
Галогенирование карбоновых кислот по
Гелю-фольгарду-зелинскому.
Эта реакция является примером взаимодействия по a-углеродному
атому по отношению к карбоксильной группе. Реакция бромирования протекает
энергично и с хорошим выходом в присутствии небольших количеств фосфора.
RCH2COOH
+ Br2
RCHBrCOOH + HBr
Функция фосфора состоит в образовании трехбромистого
фосфора, который, реагируя с кислотой, дает ацилбромид. Последний гораздо легче
подвергается енолизации по сравнению с исходной кислотой. Бром по видимому
реагирует с енолом ацилбромида так же как с енолами кетонов.
2P + 3Br2
® 2PBr3
RCH2COOH
+ PBr3 ® RCH2COBr
+ POBr + HBr
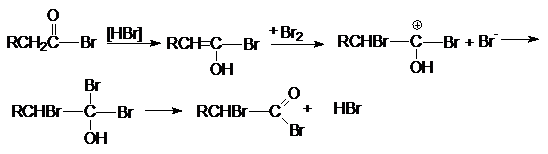
Регенерация исходного ацилбромида осуществляется за счет
взаимодействия полученного монобромацилбромида с исходной кислотой.
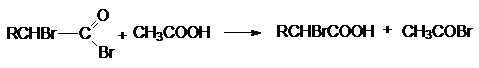
Хлор в присутствии следов фосфора реагирует аналогичным
образом, но в целом менее селективно, поскольку конкурентно может происходить
свободнорадикальное хлорирование во все положения углеродной цепи.
Взаимодействие с электрофильными реагентами.
Атомы кислорода карбонильной группы недостаточно заряжены,
чтобы быть объектом электрофильной атаки со стороны слабых и умеренных
электрофильных реагентов. Для осуществления эффективной реакции электрофильного
замещения по карбонильному углероду карбоксильной группы активируют, переводя
ее карбоксилат – ионную форму. Тогда можно осуществить реакцию алкилирование,
ацилирования и др.

Реакции восстановления карбоновых кислот.
В целом карбоновые кислоты с трудом поддаются
восстановлению как путем каталитического гидрирования, так и при действии натрия
в спирте, но восстановление до первичных спиртов при действии
литийалюминийгидрида или натрийборгидрида протекает достаточно энергично.
4RCOOH + 3LiAlH4
® [(RCH2O)4Al]Li
+ 4H2 + 2LiAlO2
[(RCH2O)4Al]Li
+ 3H2O + HCl ® RCH2OH + Al(OH)3 + LiCl
Функциональные производные карбоновых кислот можно
рассматривать как результат замещения гидроксильной группы кислоты на
какую-либо другую группу Х. Эти вещества могут быть гидролизованы в кислоту в
соответствии с уравнением.
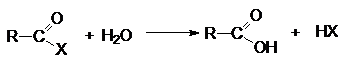
К таким соединениям относят
|
галогенангидриды |
|
|
сложные эфиры |
|
|
ангидриды |
|
|
амиды |
|
|
имиды |
|
/Image790.gif)
/Image792.gif)
/Image791.gif)
/Image794.gif)
/Image793.gif)
Общим элементам структуры этих соединений
является ацильная группа
/Image795.gif)
Однако нитрилы R-CºN также часто рассматриваются как производные карбоновых кислот,
поскольку их гидролиз приводит к карбоновым кислотам.
Галогенангидриды
карбоновых кислот.
Соединения этого ряда выражаются формулой:
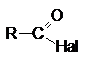
1.
Взаимодействие
карбоновых кислот с галогенангидридами неорганических кислот (SOCl2,
SO2Cl2, PCl3, POCl3, PCl5,
PBr5, COCl2)
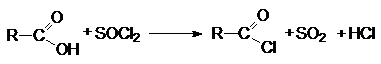
Так получают бромиды и хлориды.
2.
Реакции
диспропорционирования с галогенангидридами органических кислот (оксалилхлорид,
бензоилхлорид).
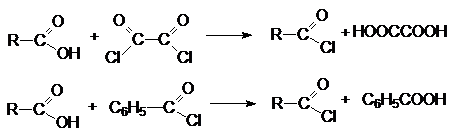
Эти реакции лежат в основе синтеза ацилхлоридов.
Физические
свойства и строение.
Ацилгалогениды представляют собой бесцветные жидкости или
кристаллические вещества с острым запахом, легколетучие, на воздухе
"дымят". Низшие ацилфториды газообразны. В воде растворяются плохо,
но быстро с ней реагируют. В ацильной группе заряд на карбонильном углероде
существенно завышен по сравнению с карбонильным углеродом карбоновых кислот
из-за сильных электроакцепторных свойств атомов галогенов
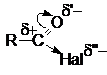
Это обусловливает высокую склонность галогенангидридов к
взаимодействию с нуклеофильными реагентами.
Реакции нуклеофильного замещения.
Ацилгалогениды легко реагируют с различными нуклеофильными
реагентами, причем эти реакции не требуют катализа. Общая схема этих реакций
может быть представлена уравнениями
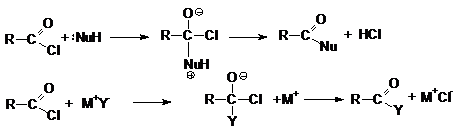
Важными реакциями нуклеофильного замещения по ацильному
углероду являются:
1.
Гидролиз
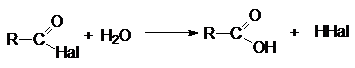
2. Взаимодействие со спиртами (этерификация)
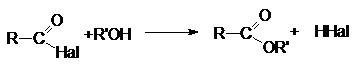
3. Взаимодействие с аммиаком и аминами (аммонолиз)
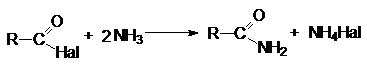
4. Взаимодействие с гидразином и его
производными
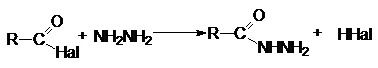
1. Восстановление
натрийборгидридом лития или литийалюминий гидридом.
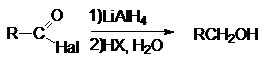
2. Восстановление
по Розенмунду
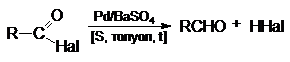
Реакция с диазометаном
(реакция Арндта-Эйстерта)
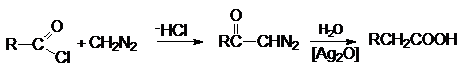
Эта реакция является синтетическим средством увеличения
длины углеводородных цепей органических соединений на один атом углерода.
Строение ангидридов выражается следующей общей формулой
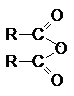
1. Термическая дегидратация карбоновых
кислот.
2RCOOH (RCO)2O
+ H2O
2. Взаимодействие с водоотнимающими агентами.
2RCOOH (RCO)2O
+ 2HPO3
2RCOOH (RCO)2O
+ 2CF3COOH
3. Взаимодействие ацилгалогенидов с солями
карбоновых кислот.
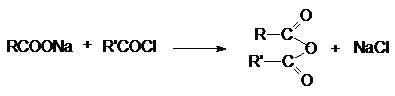
4. Взаимодействие кетена и карбоновых кислот.
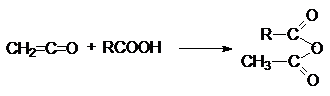
Химические
свойства ангидридов карбоновых кислот
Одна ацильная группа действует на другую как сильный
акцептор, поэтому на карбонильном атоме углерода концентрируется достаточно
высокий положительный заряд. Это обусловливает высокую реакционную способность
ангидридов в реакциях нуклеофильного замещения
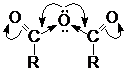
Реакция нуклеофильного замещения.
Ангидриды карбоновых кислот легко реагируют с различными
нуклеофильными реагентами, хотя скорость реакции меньше, чем в случае ацилгалогенидов.
Общий механизм реакции ангидридов карбоновых кислот с
нуклеофилом может быть представлен схемой:
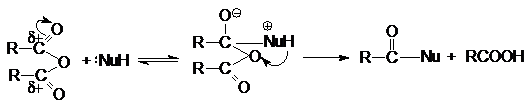
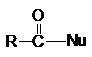 Можно видеть, что в результате реакции
кроме продукта нуклеофильного
Можно видеть, что в результате реакции
кроме продукта нуклеофильного
замещения
образуется карбоновая кислота.
Важнейшими реакциями нуклеофильного замещения с участием
ангидридов карбоновых кислот является:
1. Гидратация.
(RCO)2O + H2O ® 2RCOOH
2. Этерификация.
(RCO)2O + R'OH ® RCOOR' + RCOOH
3. Взаимодействие с аммиаком и аминами
(аммонолиз).
(RCO)2O
+ NH3 ® RCONH2
+ RCOOH
Кетены.
Кетен формально может рассматриваться как ангидрид уксусной
кислоты.
CH2=C=O
+ H2O ® CH3CHO
Кетен получают следующими методами:
1. Пиролизом кетонов или карбоновых кислот на
фосфатных катализаторах.
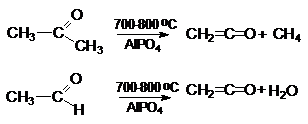
2. Отщепление галогеноводорода от
ацилхлоридов в присутствии сильного органического основания, например,
третичного амина.
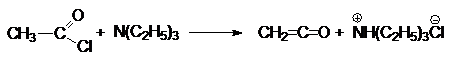
3. Дегалогенирование a-
галогенацилгалогенида
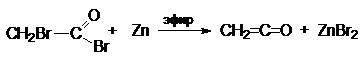
![]() Химическое поведение кетена определяют два
фактора: напряженность
Химическое поведение кетена определяют два
фактора: напряженность
молекулы и высокий положительный заряд на карбонильном
углероде
Наиболее типичными реакциями кетенов являются реакции
нуклеофильного присоединения.
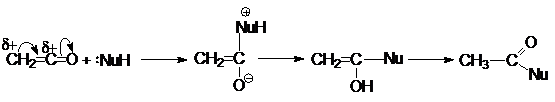
Наиболее важными среди этих являются реакции гидратации,
образования ангидридов сложных эфиров и амидов карбоновых кислот:
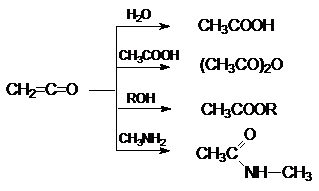
Поскольку в результате этих реакций не образуется
каких-либо других продуктов, то кетен является идеальным ацилирующим агентом.
Сложные эфиры – производные карбоновых кислот, которые
можно рассматривать как результат замещения гидроксильной группы карбоновой кислоты
на алкоксирадикал
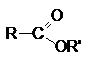
Методы
получения сложных эфиров.
1. Этерификация карбоновых кислот (см.
химические свойства карбоновых кислот).

2. Ацилирование спиртов галогенангидридами и
ангидридами карбоновых кислот.
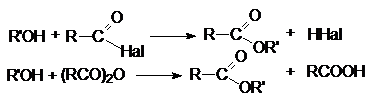
3. Ацилирование алкоголятов
галогенангидридами и ангидридами карбоновых кислот.
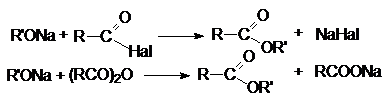
4. Алкилирование солей карбоновых кислот.
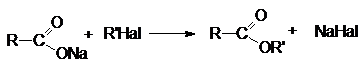
5. Этерификация амидов и нитрилов карбоновых
кислот.
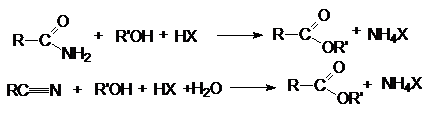
6.
Реакция
Байера-Виллигера

Методы
синтеза лактонов (циклических сложных эфиров).
1. Внутренняя этерификация g- и s-
оксикарбоновых кислот.
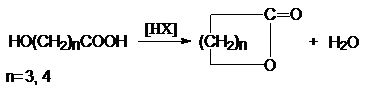
Физические свойства
и строение сложных эфиров.
Сложные эфиры являются бесцветными жидкостями или
кристаллическими веществами с приятным запахом. Температура кипения сложных
эфиров обычно ниже чем температура близких по молекулярной массе карбоновых
кислот. Это свидетельствует об уменьшении межмолекулярных взаимодействий, что
объясняется отсутствием межмолекулярных водородных связей.
Полярность связей в молекуле сложного эфира подобна
полярности связи в карбоновых кислотах.
Основное отличие от карбоновых кислот – отсутствие
подвижного протона, вместо него находится углеводородный остаток. Электрофильный
центр находится на карбонильном и алкильном углеродном атоме. В то же время
карбонильный кислород обладает основностью.
Объектами нуклеофильной атаки могут быть ацильный или
алкильный углерод. В то же время кислотность водородных атомов при a-углеродном
атоме радикала кислоты обусловливает склонность сложность эфиров к реакции
конденсации.
Реакции нуклеофильного замещения.
1. Гидролиз.
Различают кислотнокаталитический гидролиз
и основной гидролиз (омыление). Кислотный гидролиз представляет собой обратимую
реакцию.
RCOOR'
+ H2O ![]() RCOOH + R'OH
RCOOH + R'OH
Механизм этой реакции – см в разделе
Кислотно-каталитическая этерификация – Химические свойства карбоновых кислот.
RCOOR'
+ NaOH ® RCOONa
+ R'OH
2. Аммонолиз.
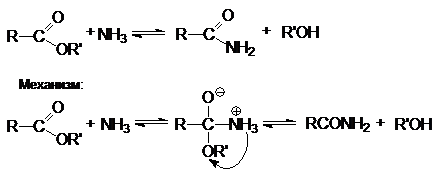
3.
Переэтерификация.
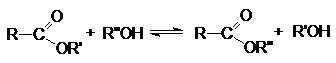
Реакция катализируется кислотами или основаниями.

Реакции восстановления.
Синтезы на
основе эфиров 1,3-кетокислот.
Кето-енольная таутомерия эфиров 1,3-кетокислот и реакции
таутомерных форм.
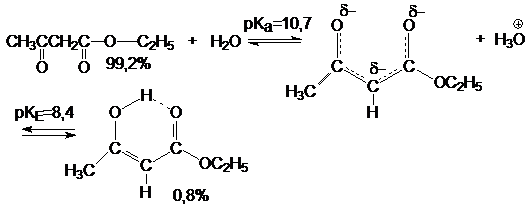
Эфиры 1,3-кетокислот по своему химическому поведению близки
к 1,3-дикетонам. Однако их кислотность ниже. Для них характерна кетоенольная таутомерия.
Как и в случае 1,3-дикетонов таутомерное равновесие
чувствительно к сольватирующей способности реакционной среды. Так в растворе
метанола образуется 8,7% енола.
Анионы эфиров 1,3-кетокислот представляют собой сопряженную
систему с выровненными связями и делокализованным отрицательным зарядом. Они содержат несколько
реакционных центров. Их соли в растворах существуют в виде ионных пар. С ионами
тяжелых металлов они образуют внутренние комплексы (хелаты). Соли
1,3-кетокислот подобны солям 1,3-дикетонов. Они легко алкилируются и
ацилируются
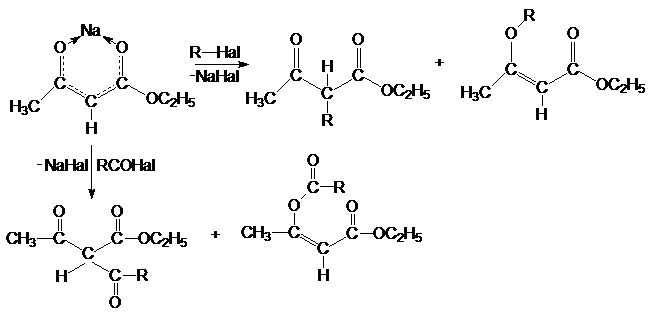
2. Ацилоиновая конденсация.
В отличие от конденсации Кляйзена в ацилоиновой конденсации
ключевую роль играют сложноэфирные группы. Суммарно ацилоиновая конденсация
может быть представлена схемой:
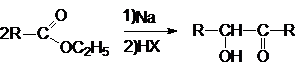
Эти соединения следует рассматривать как результат
замещения гидрокси-группы в карбоновой кислоте на амино-группу
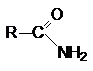
1. ацилирование аммиака и аминов.
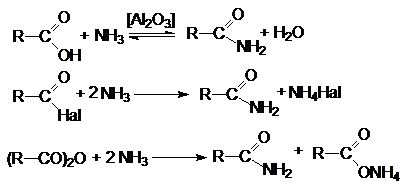
2. Дегидратация аммониевых солей карбоновых
кислот
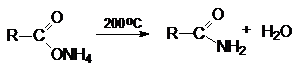
3. Гидролиз нитрилов карбоновых кислот
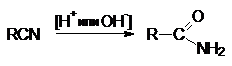
Синтез лактамов – циклических амидов.
1. Перегруппировка циклических амидов –
лактамов
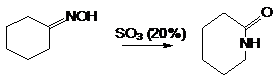
2. Реакция Байера-Виллигера
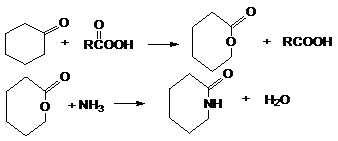
3. Циклизация аминокислот (n³3)
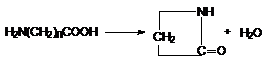
Физические
свойства амидов. Строение амидной группы.
Амиды представляют собой бесцветные кристаллические
вещества или жидкости, растворяющиеся в органических растворителях. Амиды в
молекулах которых имеются связи N-H
ассоциированы вследствие образования межмолекулярных водородных связей и имеют
более высокие температуры кипения.
В молекулах амидов имеет место значительное сопряжение неподеленной парой электронов азота и p-электронной системой двойной связи С=О. Это приводит к образованию дополнительной поляризации связей в амидной группе и наличие электрофильных реакционных центров на ацильном и алкильных углеродных атомах и отрицательного – на карбонильном кислороде.
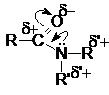
Реакции нуклеофильного замещения.
Примером может служить гидролиз. В нейтральной среде
гидролиз протекает медленно. Поэтому реакцию ведут в присутствии минеральной
кислоты либо основания, которые не только ускоряют ее, но и участвуют как реагенты.
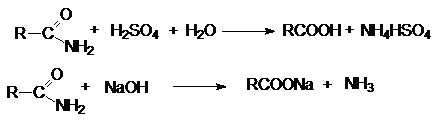
Активация молекулы амида кислотой связана с ее
протонированием и увеличением положительного заряда на карбонильном углероде,
который становится более восприимчивым к последующей нуклеофильной атаке:
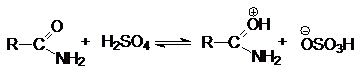
RCONH2 RCH2NH2
RCONHR' RCH2NHR'
RCONHR'2
RCH2NHR'2
1. Реакция
дегидратации.
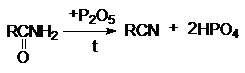
Расщепление по Гофману.
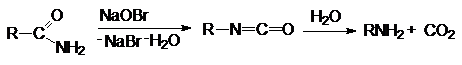
2.
Взаимодействие формамидов с реактивом
Гриньяра (реакция Буво)
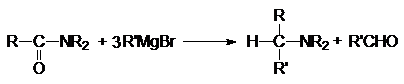
Продуктами этой реакции являются альдeгид и третичный амин.
Общая формула нитрилов R-CºN или CnH2n+1CN.
1. Дегидратация амидов карбоновых кислот с
помощью водоотнимающих агентов.

На практике в последнем случае пропускают над катализатором
смесь карбоновой кислоты с аммиаком. В этом процессе совмещается образование
амида и его дегидратация.

2. Нуклеофильное замещение галогена в
галогеналканах на циaнид-анион.
R-Hal + NaCN![]() R-CºN + NaHal
R-CºN + NaHal
Циано-группа характеризуется высокой степенью поляризации,
следствием которой является образование частичного положительного заряда на
углероде и отрицательного заряда – на азоте
![]()
Это обусловливает, с одной стороны, восприимчивость
углеродного центра к нуклеофильной атаке, с другой – основность атома азота.
1. Реакции
нуклеофильного присоединения.
Нитрилы легко реагируют с сильными анионными нуклеофильными
реагентами (карб-анионами, амидами металлов, щелочами, алкоголятами, тиоалкоголятами)

Примером может служить щелочной гидролиз нитрилов:
Дальнейший гидролиз амида приводит к образованию солей
карбоновых кислот.
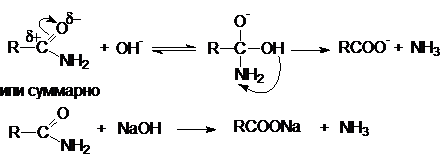
Таким образом, суммарное уравнение щелочного гидролиза
нитрилов можно представить как
R-CºN + H2O
+ NaOH ® RCOONa
+ NH3
Реакции нитрилов со слабыми нуклеофилами (водой, спиртами)
протекает крайне медленно. Для эффективного проведения этих реакций используют
кислотный катализ. Активация молекулы нитрила в этом случае осуществляется за
счет образования водородной связи между кислотой – катализатором и атомом азота
и, как следствие, увеличение положительного заряда на нитрильном углеродном
атоме.
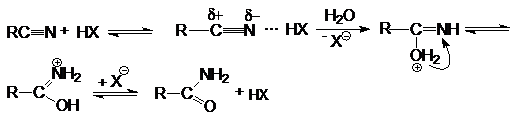
Таким образом, полный кислотный гидролиз нитрилов
описывается уравнением:
R-CºN + 2H2O + HX ® RCOOH + NH4X
Если в качестве реагента использовать водно-спиртовые
среды, то можно осуществлять синтез сложных эфиров
R-CºN + R'OH + H2O + HX ® RCOOR' + NH4X
2.
Гидрирование.
Реакции гидрирования нитрилов можно подразделить на две
группы:
а) каталитическое гидрирование
RCºH RCH2NH2
В качестве катализаторов этих реакций используют
металлические Pt, Pd, Ni.
б) реагентное гидрирование
RCºH ![]() ®RCHO + NH3 + LiX
®RCHO + NH3 + LiX
В качестве реагентов этих реакциях используют LiAlH4, NaBH4.
Угольную кислоту формально можно рассмотреть как
карбоновую кислоту, которая вместо углеводородого остатка содержит гидроксильную
группу Cвойства производных угольной кислоты в основном подобны
свойствам производных карбоновых кислот.
Отличие кроется в том, что производные угольной
кислоты представляют собой результат замещения одной или двух гидроксильных
групп.
Поэтому
и те и другие являются бифункциональными соединениями. Это открывает
допонительные возможности вариации их структуры, а также делает симметричные
структуры потенциальным сырьем для получения поликонденсационных полимеров.
Рассмотрим некоторые наиболее важные производные
угольной кислоты.
 Фосген хлоругольная кислота
Фосген хлоругольная кислота
Фосген является устойчивым
соединением, хлоругольная кислота неустойчива, известны ее производные, например
эфиры.
Фосген получают свободнорадикальным хлорированием
оксида углерода (II)
CO + Cl2 COCl2
Фосген и эфиры хлоругольной кислоты проявляют свойства хлорангидридов карбоновых кислот, однако в отличие от последних более реакционноспособны в реакциях нуклеофильного замещения. Они являются реагентами для получения эфиров угольной и хлоругольной кислот.
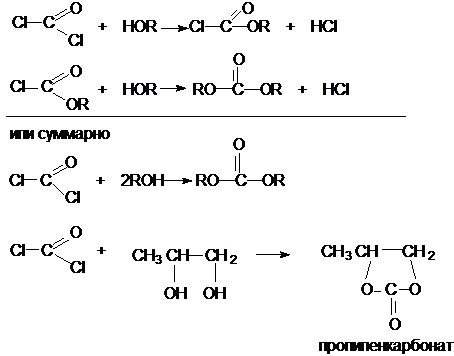
Если
в качестве реагента используют фенолы, то результатом является образование
диарилкарбонатов.
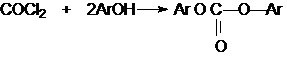
Фосген
является бифункциональным соединением, поэтому его используют для получения
пластмасс – поликарбонатов.
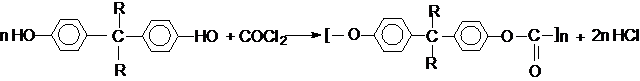
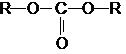
Эти соединения проявляют свойства обычных эфиров
карбоновых кислот, в том числе вступают в реакции сложноэфирной конденсации и
поэтому используются в органическом синтезе для введения в структуру органических
алкоксикарбонильной группы.
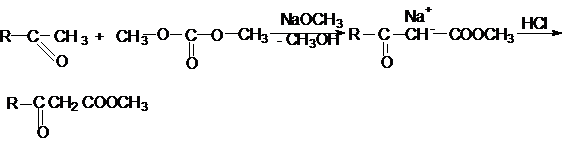
Типичным представителем амидов угольной кислоты
является мочевина (карбамид)
![]()
В промышленности она может быть получена из аммиака и CO2
Замещенные мочевины могут быть получены взаимодействием
фосгена с аминами
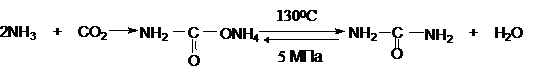
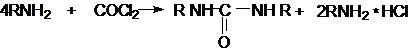
Эта реакция осуществляется ступенчато через промежуточное образование
карбаминоилхлорида

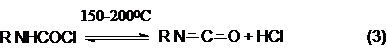
Можно видеть, что синтез замещенных мочевин требует
применение избытка амина. Если реакцию проводить в избытке фосгена , то
количественно образуется карбаминоил хлорид (реакция (1)). Последний может быть
использован для получения изоцианатов:
![]()
причем
реакцию проводят в условиях диссоциации образующегося гидрохлорида амина,
образующегося на стадии (1) и вступающего
в реакцию (4).
![]()
Суммируя реакции (1), (3), (4), имеем стехиометрию
процесса синтеза изоцианата
![]()
Изоцианаты используют для получения
уретанов (эфиров карбаминовой кислоты)
![]()
Cами карбаминовые кислоты RNHCOOH ,
представляющие собой алкиламиды нестабильны и легко распадаются на
амины(аммиак) и CO2
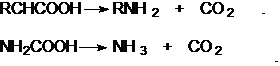
Двухосновные, ненасыщенные и аренкарбоновые кислоты. Дикарбоновые кислоты
Эти соединения могут рассматриваться как результат замещения двух атомов
водорода в углеводородах на карбоксильные группы.
Дикарбоновые кислоты можно подразделить на три группы, в зависимости от
характера углеводородного остатка – насыщенные, ненасыщенные и арендикарбоновые
кислоты.
Методы
получения насыщенных дикарбоновых кислот.
1. Окисление a,w-диолов.
HO(CH2)nOH HOOC(CH2)n-2COOH
В качестве окислителей можно использовать KMnO4 в
нейтральной или кислой среде или K2Cr2O7 в кислой среде.
2. Окисление циклоалканов.
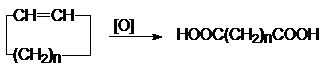
Реакцию проводят при повышенных температурах, используя в качестве окислителей
KMnO4 или K2Cr2O7 в кислой
среде.
Кислотные свойства
дикарбоновых кислот.
Каждая карбоксильная группа действует на другую как
электроноакцептор, повышая ее кислотность, причем с увеличением расстояния
между карбоксильными группами их кислотность падает.
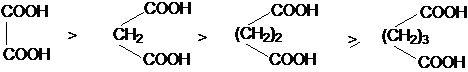
В силу быстрого затухания индуктивного эффекта
различие в кислотностях дикарбоновых кислот с ростом длины полиметиленовой кислоты
возрастает.
Вторая константа кислотности дикарбоновых кислот ниже,
чем первая, поскольку карбоксилат-анион обладает +I – эффектом.
Наиболее важными среди дикарбоновых кислот являются щавелевая, малоновая,
янтарная, глутаровая (этан-, пропан-, бутан- и пентандикарбоновая).
Щавелевая кислота – НООС-СООН, бесцветное кристаллическое вещество, растворимое в воде и
спиртах и труднорастворимое в углеводородах.
Специфическим способом получения щавелевой кислоты является нагревание формиата натрия.
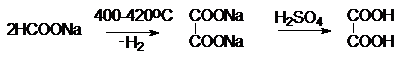
Специфическими реакциями щавелевой кислоты являются разложение в присутствии
концентрированной серной кислоты и окисление:
HOOC-COOH CO +CO2
+H2O
HOOC-COOH CO2 +H2O
Щавелевую кислоту используют как протраву при крашении тканей и как
аналитический агент. Возможно ее использование для получения полиэфиров. Из производных
щавелевой кислоты интерес представляет диэтилоксалат С2Н5ООС-СООС2Н5.
Для него характерны многие реакции сложных эфиров. Он вступает в реакцию
сложноэфирной конденсации (см. конденсацию по Клайзену) в качестве реагента, не
содержащего подвижные атомы водорода (см. синтезы на основе ацетоуксусного
эфира). Подобный же вид конденсации протекает при взаимодействии диэтилоксалата
с кетонами.
Малоновая кислота.
НООС(СН2)СООН – бесцветное кристаллическое вещество,
растворимое в воде.
Специфичный способ получения – из хлоруксусной кислоты
ClCH2COONa NCCH2COONa
HOOC(CH2)COOH
Малоновая кислота легко декарбоксилируется при нагревании выше 133-135оС
или при кипячении водных растворов в присутствии кислот.
HOOCCH2COOH CH3COOH + CO2
Малоновая кислота содержит очень активную метиленовую группу с подвижными
атомами водорода. Поэтому малоновая кислота и ее эфиры вступают в реакции
конденсации, протекающие по карбанионному механизму:
а) конденсация с альдегидами и кетонами (реакция Кневенагеля):
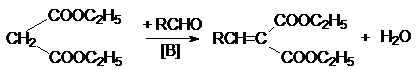
б) реакция Михаэля – сопряженное присоединение к a,b-ненасыщенным системам:
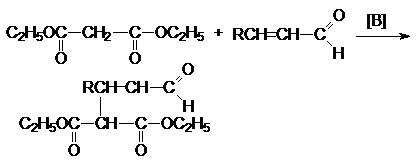
Янтарная кислота – HOOC(CH2)2COOH – бесцветное кристаллическое вещество, растворимое в
воде и спиртах.
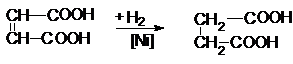
В промышленности ее получают гидрированием малеиновой
кислоты и карбонилированием этиленгликоля
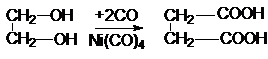
Для янтарной кислоты характерны все реакции
карбоксильной группы. Особенность янтарной кислоты – способность к циклизации.
Наиболее важными реакциями циклизации являются образование янтарного ангидрида
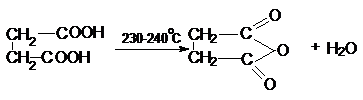
и сукцинимида:
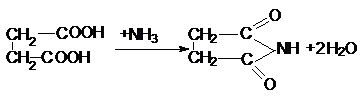
Адипиновая кислота.
Получается окислением циклогексанона азотной кислотой:
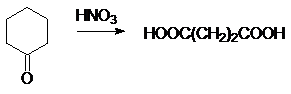
и карбонилированием 1,4-бутандиола и тетрагидрофурана:
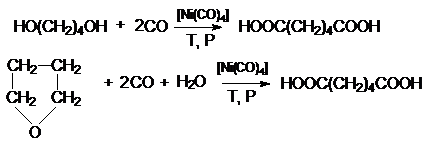
Адипиновая кислота используется для получения волокна
найлон.
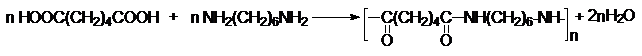
Эфиры адипиновой кислоты и ее ближайших гомологов
используются для получения средних и
больших циклов. В этом плане важными синтезами являются:
Ацилоиновая конденсация:
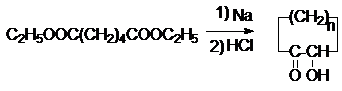
Конденсация по Дикману:

Механизм
последней реакции подобен механизму конденсации по Кляйзену
a,b-Непредельные
карбоновые кислоты.
1)
Дегидратация b-оксикислот
R-CH(OH)CH2COOH
R-CH=CH-COOH
2) Дегидрогалогенирование галогенкарбоновых кислот
R-CH(Hal)-CH2COOH
R-CH=CH-COOH +
NaHal + H2O
R-CH(Hal)-CH(Hal)-COOH
R-CºC-COOH + 2NaHal +
2H2O
3)
Гидрокарбоксилирование алкинов
HCºCH + CO + H2O
CH2=CH-COOH
4) Окисление a,b-ненасыщенных спиртов или альдегидов в мягких условиях
RCH=CH-CHO RCH=CH-COOH
RCH=CH-CH2OH
RCH=CHCOOH
Окисление осуществляtтся кислородом при катализе
солями кобальта и марганца или неорганическими окислителями (K2Cr2O7 в кислой среде, KMnO4 в кислой
или нейтральной среде)
Подобно алкенам двойные
связи a,b-ненасыщенных кислот могут присоединять бром, эпоксидироваться
, гидробромироваться , хотя эти реакции оказываются более медленными по
сравнению с соответствующими реакциями алкенов, что связано с дезактивирующим
действием карбоксильной группы.
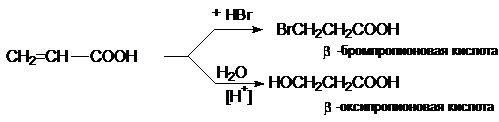
Присоединение воды, HBr и других реагентов, имеющих подвижный водород, идет
против правила Марковникова.
Ненасыщенные дикарбоновые кислоты.
Важнейшими из соединений этого ряда являются
малеиновая кислота и фумаровая кислота, которые относятся друг к другу как цис- и транс- изомеры.
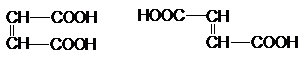
Малеиновая кислота получается каталитическим
парофазным окисление бензола и бутена –2.

Классификация,
номенклатура, изомерия.
Аренкарбоновые кислоты содержат карбоксильную группу в ароматическом
кольце или боковой цепи. По количеству карбоксильных групп различают моно-, ди-
и поликарбоновые кислоты. Примеры номенклатуры аренкарбоновых кислот и их замещенных
приведены в таблице.
1) Реакции
окисления
Аренкарбоновые кислоты, содержащие карбоксильую
группу, связанную с ароматическим кольцом, получают окислением алкиларенов.
ArCH3 ArCOOH
В качестве окислителей используют KMnO4, Na2Cr2O7,
кислород в присутствии солей Co и Mn.
Метилкетоны также могут быть окислены в кислоты, лучше всего действием галогенов в щелочной среде (галоформная реакция):
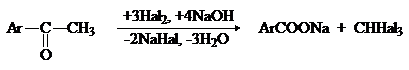
2) Реакции
гидролиза
Наиболее широко используют гидролиз нитрилов, которые
могут быть получены из галогенаренов или солей арилдиазония:
ArCN ArCOOH
Кроме того, для синтеза аренкарбоновых кислот могут быть использованы
карбоксилирование магний- и литийорганических соединений, а также непосредственное
введение карбоксильной группы в ароматическое кольцо - карбоксилирование
фенолятов (см. лекцию №29).
Физические
свойства и строение.
Аренмонокарбоновые кислоты – бесцветные кристаллические вещества.
Бензольное кольцо и карбоксильная группа образуют сопряженную систему. Карбоксильная группа действует как электроноакцептор (-I и –M-эффекты).
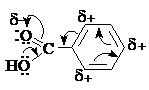
Реакции аренкарбоновых кислот по карбоксильной группе аналогичны соответствующим
превращениям алифатических карбоновых кислот.
Кислотные свойства
Аренкарбоновые кислоты превышают по кислотности насыщенные и a,b-непредельные карбоновые кислоты. На силу аренкарбоновых кислот влияет
природа и положение заместителя в ароматическом кольце. Это влияние согласуется
с электронными эффектами заместителей.
Значения рКа
для замещенных бензойных кислот X-C6H4-COOH.
|
X |
CH3 |
F |
Cl |
Br |
I |
OH |
OCH3 |
NO2 |
|
Положение |
|
|||||||
|
мета |
4,24 |
3,86 |
3,83 |
3,81 |
3,86 |
4,08 |
4,09 |
3,45 |
|
пара |
4,34 |
4,14 |
3,99 |
4,00 |
3,92 |
4,58 |
4,47 |
3,43 |
Для бензойной кислоты рКа=4,17.
Таким образом, заместители, проявляющие –М- и –I-эффекты (NO2), увеличивают кислотность, причем из пара-положения в большей степени, чем из
мета-положения. Заместители с +М-эффектом,
превышающим –I-эффект (OH, OCH3), из пара-положения
уменьшают, а из мета-положения увеличивают
кислотность. Заместители с –I-эффектом,
превышающим +М-эффект (галогены), увеличивают кислотность, причем из мета-положения в большей степени, чем из
пара-положения.
При оценке влияние положения заместителя на кислотность следует иметь в
виду, что пара-положение находится в
сопряжении с карбоксильной группой, что обеспечивает влияние мезомерного
эффекта заместителя, находящегося в этом положении, на кислотный центр. Из мета-положения М-эффект заместителя
передается только на атомы углерода ароматического кольца.
Реакции замещения в ароматическом кольце
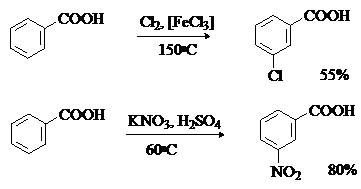
Аренкарбоновые кислоты вступают в реакции с электрофилами с образованием
продуктов электрофильного замещения (нитрования, сульфирования, галогенирования)
в бензольном кольце. Реакции протекают в жестких условиях, заместитель вступает
преимущественно в мета-положение к
карбоксильной группе.
Основной метод получения – окисление диметиларенов.
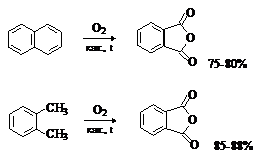
Фталевую кислоту в виде ангидрида в промышленности получают окислением
о-ксилола или нафталина кислородом воздуха в присутствии катализаторов на основе
V2O5-TiO2 при 350-4000С:
Терефталевую кислоту получают окислением п-ксилола кислородом в уксусной
кислоте в присутствии солей Co и Mn:
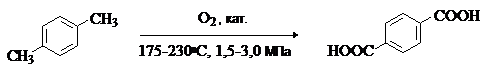
Другой промышленный метод получения терефталевой кислоты – изомеризация дикалиевой соли о-фталевой кислоты:
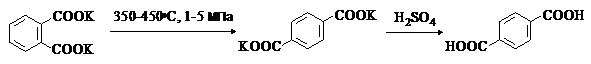
Химические свойства арендикарбоновых кислот в основном аналогичны свойствам
монокарбоновых кислот. Они дают все реакции, характерные для карбоксильной
группы. Арендикарбоновые кислоты по первой ступени являются более сильными
кислотами, чем монокарбоновые:
|
|
рКа1 |
рКа2 |
|
Бензойная кислота |
4,17 |
- |
|
Фталевая кислота |
2,95 |
5,41 |
|
Изофтелевая кислота |
3,46 |
4,46 |
|
Терефталевая кислота |
3,51 |
4,82 |
Фталевая кислота и ее производные
Фталевая кислота – бесцветное кристаллическое вещество, мало растворима в воде. При нагревании в присутствии дегидратирующих агентов легко превращается во фталевый ангидрид:
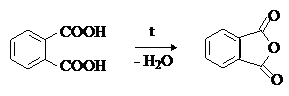
Практическое применение имеют производные фталевой кислоты – ангидрид,
имид, сложные эфиры.
Фталевый ангидрид – бесцветное кристаллическое вещество, легко возгоняется.
При взаимодействии со спиртами в присутствии серной кислоты дает сложные моно- и диэфиры:
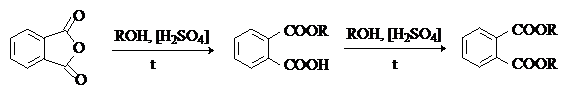
Диалкиловые эфиры фталевой кислоты (диалкилфталаты) используют как пластификаторы
полимеров, высококипящие растворители, репелленты.
С многоатомными спиртами фталевый ангидрид образует полиэфиры. Например, на основе фталевого ангидрида и глицерина получают алкидные (глифталевые) смолы:
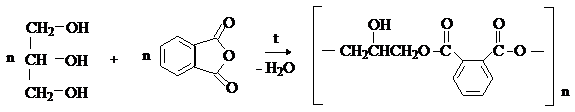
С аммиаком и первичными аминами фталевый ангидрид в зависимости от условий
дает фталиминовую кислоту, фталимид или диамид фталевой кислоты:
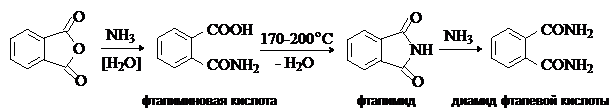
Ацилирование фталевым ангидридом бензола и алкилбензолов приводит к
о-ароилбензойным кислотам и далее к антрахинонам:
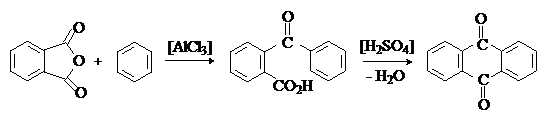
При конденсации фталевого ангидрида с фенолами образуются производные
трифенилметана, которые называют фталеинамина. Их используют как индикаторы и
красители (фенолфталеин, флуоресцеин):

Фталимид – бесцветное кристаллическое вещество, легко
возгоняется.
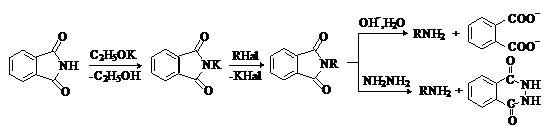
Фталимид является NH-кислотой (рКа=9,9).
Он растворяется в водных растворах щелочей. При этом происходит его постепенный
гидролиз с раскрытием цикла и образованием фталиминовой кислоты. В безводных
средах под действием оснований образуются соли фталимида, которые используются
в органическом синтезе для получения первичных аминов (синтез Габриэля):
Терефталевая кислота – бесцветное кристаллическое вещество, мало растворима
в воде и органических растворителях. Крупнотоннажный промышленный продукт. Её
диметиловый эфир (диметилтерефталат) используется для получения полиэтилентерефталата, который применяют
для изготовления синтетического волокна лавсан: