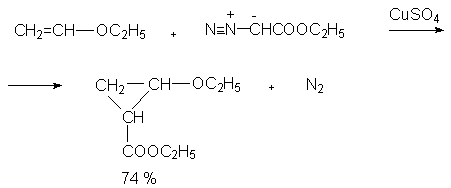Амины и
нитросоединения, свойства, методы получения.
Нитросоединения – производные углеводородов в которых
один или несколько атомов водорода замещены нитрогруппой – NO2.
Нитроалканы – производные алканов, в которых один или
несколько атомов водорода замещены нитрогруппой.
Общая формула мононитроалканов CnH2n+1NO2.
При образовании названий нитроалканов выбирается самая
длинная углеводородная цепь, нумерация которой начинается с конца, к которому
ближе расположена нитрогруппа. Последняя указывается с помощью приставки
«нитро». Например:
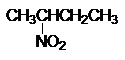 2-нитробутан
2-нитробутан
1.Нитрование
алканов
RH RNO2
Из метана получают нитрометан, при нитровании
гомологов метана образуется смесь нитроалканов:
CH3CH2CH3
CH3NO2
+ CH3CH2NO2 + CH3CH(NO2)CH3
+ CH3CH2CH2NO2
2.Алкилирование нитритов
R-Br + AgNO2
® R-NO2 + AgBr
R-Br + NaNO2
® R-NO2 + NaBr
Поскольку нитрит-анионы имеют амбидентный характер,
для получения высокого выхода нитроалкана используют апротонные неполярные
растворители и умеренные температуры.
Физические свойства и строение.
Нитроалканы являются бесцветными или желтоватыми
жидкостями или кристаллическими
веществами со слабым запахом.
Для мононитроалканов характерны большие дипольные
моменты. Причиной значительной полярности нитроалканов кроется в электронном
строении нитрогруппы, содержащей семиполярную связь
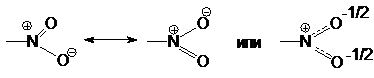
Выравнивание связей N-O подтверждается
рентгеноструктурным анализом: связь N-O в нитрогруппе короче связи N-O в гидроксиламине,
но длинее связи в нитрозогруппе –N=O.
Высокая электроотрицательность атомов N и О, кратность
связи N=O и ее семиполярный
характер обусловливают значительные электроноакцепторные свойства нитрогруппы (-I и –М-эффекты).
Для нитроалканов характерно слабое поглощение в
УФ-области 270-280 нм. Это связано с электронными переходами типа p®p* неподеленной
электронной пары атома кислорода на НСМО.
В ИК-спектрах наблюдаются максимумы поглощения
связанные с симметричными и
антисимметричными колебаниями связей N=O в областях 1370
см-1 и 1550 см-1.
Химические свойства нитроалканов.
Кислотность и
таутомерные превращения нитроалканов.
Первичные и вторичные нитроалканы являются СН- кислотами.
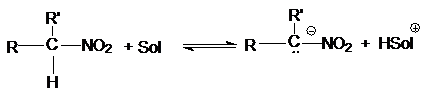
Кислотность обусловлена стабилизацией образующегося
карбаниона за счет электроноакцепторных свойств нитрогруппы.
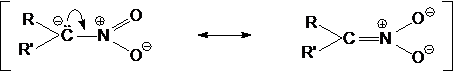
Кислотность мононитроалканов в водных растворах
сравнима с кислотностью фенолов. Если у одного атома углерода находится две или
три нитрогруппы, кислотность резко возрастает.
Анион нитроалакана является амбидентным подобно
енолят-аниону. Например, при его протонировании может образовываться, кроме
нитроалкана, другая таутомерная форма.
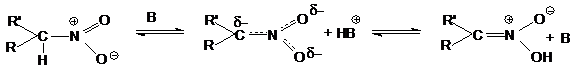
Таутомерную форму нитроалкана называю ациформой или
нитроновой кислотой, которая в чистом виде не получена. Нитроновая кислота
является ОН- кислотой средней силы (рКа=3,2).
Таким образом, нитросоединения следует рассматривать
как таутомеры, реагирующие в нитро- и аци-формах.
В обычных условиях концентрация аци-формы ничтожна (10-5-10-7%).
Равновесие смещается в правую сторону в щелочной среде вследствие образования солей.
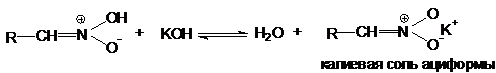
Кристаллические соли щелочных и щелочно-земельных
металлов устойчивы и хорошо растворимы в воде. Их иногда называют солями
нитроновой кислоты. При подкислении растворов сначала образуется сама
нитроновая кислота (ациформа), которая затем изомеризуется в нитроалкан.
Нитросоединения относятся к псевдокислотам, для
которых характерно, что сами они нейтральны, не обладают электропроводностью,
но тем не менее образуют нейтральные соли щелочных и щелочно-земельных
металлов.
«Нейтрализация» нитросоединений основаниями происходит
медленно, а истинных кислот – мгновенно.
Из других реакций нитроалканов отметим следующие.
Гидролиз в
кислой среде с разрывом связей C-N.
CH3NO2
+ H2O HCOOH + NH2OH + H2SO4
Эта реакция используется в технике для синтеза
гидроксиламина и его сульфата.
Замещение
Н-атомов при a-С на галогены, остатки азотистой кислоты, альдегидов,
кетонов и т.д.
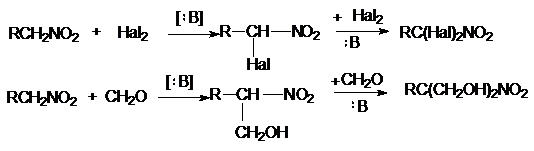
Реакция с HNO2 является качественной на нитроалканы. Третичные нитроалканы
не реагируют, вторичные R2CH-NO2 образуют нитрозонитроалканы
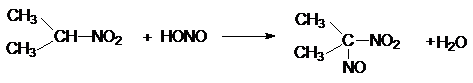
Первичные образуют с HNO2
нитрооксимы (нитроловые кислоты)
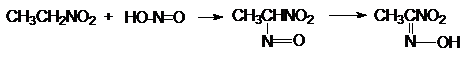
Эти бесцветные соединения образуют со щелочами соли
нитроловых кислот кроваво-красного цвета.
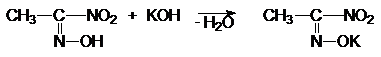
Ароматические нитросоединения.
1)
Нитрование аренов
Это основной метод получения нитроаренов; подробно
рассмотрен при изучении электрофильного ароматического замещения (см. лек.№18).
2)
Окисление ариламинов
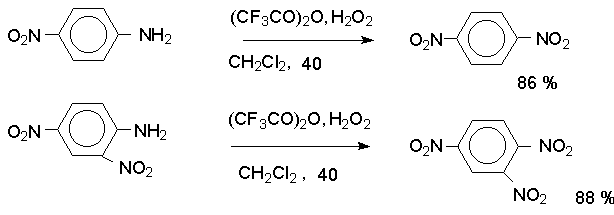
Метод
заключается в окислении первичных ароматических аминов пероксисоединениями.
Наиболее эффективным реагентом для окисления является трифторпероксиуксусная кислота в хлористом
метилене. Трифторпероксиуксусную кислоту получают непосредственно в реакционной
смеси при взаимодействии ангидрида трифторуксусной кислоты и 90%-ной перекиси
водорода. Этот метод имеет значение для синтеза нитросоединений, содержащих в орто- и пара-положениях к нитрогруппе другие электроноакцепторные группировки,
например:
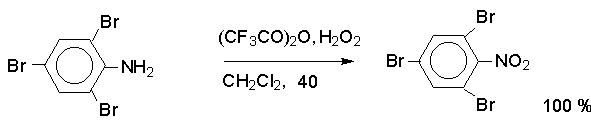
Физические свойства и строение.
Нитроарены – желтые вещества со своеобразным запахом.
Нитробензол - жидкость с запахом горького миндаля. Ди- и полинитроарены –
кристаллические вещества.
Нитрогруппа является сильным электроноакцептором,
поэтому нитроарены имеют большие дипольные моменты, направленные в сторону
нитрогруппы.
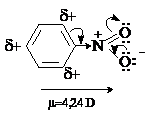
Молекулы полинитроаренов являются сильными
электроноакцепторами. Например, сродство к электрону 1,3-динитробензола составляет
1,35 эВ, а 1,3,5-тринитробензола – 1,75 эВ.
Продуктом
исчерпывающего восстановления нитрогруппы в нитроаренах являются аминогруппа. В
настоящее время для восстановления нитроаренов в ариламины в промышленных
условиях применяется каталитическое гидрирование. В качестве катализатора
используют медь на силикагеле в качестве носителя. Выход анилина над этим
катализатором составляет 98 %.

В лабораторных условиях для восстановления нитрогруппы
используют металлы в кислой или щелочной среде. Восстановление происходит в несколько
стадий, последовательность которых в кислой и щелочной среде сильно различается.
При восстановление в кислой среде
процесс протекает ступенчато и включает следующие стадии.
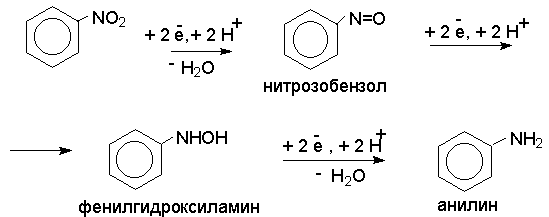
В кислой среде каждый из промежуточных продуктов
быстро восстанавливается до конечного продукта анилина и их не удается выделить
в индивидуальном виде. В качестве восстановителей применяют железо, олово или
цинк и соляную кислоту. Эффективным восстановителем нитрогруппы является хлорид
олова (II) в соляной кислоте. Конечным продуктом восстановления в кислой среде
является амин, например:
C6H5NO2 + 3Zn + 7HCl ® C6H5NH2 HCl + 3ZnCl2 + 2H2O
В нейтральном растворе, например, при восстановлении нитроаренов цинком в
водном растворе хлорида аммония, процесс восстановления замедляется и останавливается
на стадии образования арилгидроксиламина.
C6H5NO2 + 2Zn + 4NH4Cl
C6H5NHOH + 2[Zn(NH3)2]Cl2
+ H2O
При восстановлении в щелочной среде в избытке
восстановителя конечным продуктом восстановления нитроарена является гидразоарен
(диарилгидразин)
2ArNO2 +
5Zn + 10NaOH ArNHNHAr + 5Na2ZnO2
+ 2H2O
Процесс может
быть представлен в виде следующей последовательности превращений.
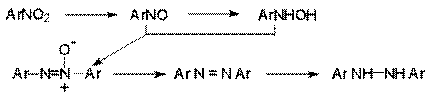
азоксиарен
азоарен гидразоарен
Скорость
процесса восстановления нитрогруппы сульфидами сильно зависит от электронных
эффектов заместителей в ароматическом кольце. Так, м-динитробензол восстанавливается дисульфидом
натрия в 1000 раз быстрее м-нитроанилина. Это используется для парциального восстановления нитрогрупп в
полинитросоединениях.
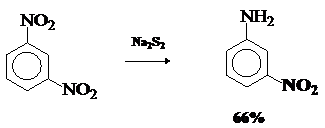
Продукты неполного восстановления нитрогруппы.
Нитрозоарены
легко восстанавливаются, поэтому их трудно получить восстановлением
нитроаренов. Лучший метод получения нитрозоаренов состоит в окислении
арилгидразинов.
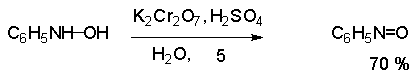
Возможно
непосредственное введение нитрозогруппы в ароматическое кольцо действием
азотистой кислоты на фенолы и третичные ариламины.
В
кристаллическом состоянии ароматические нитрозосоединения существуют в виде
бесцветных димеров. В жидком и газообразном состоянии существует равновесие
между димером и мономером. Мономеры окрашены в зеленый цвет.
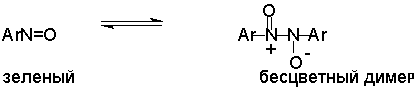
Нитрозосоединения,
подобно карбонильным, соединениям вступают в реакции с нуклеофилами. Например,
при конденсации с арилгидроксиламинами образуются азоксисоединения (см. выше),
а с ариламинами – азосоединения.
![]()
Кроме описанного выше метода получения восстановлением нитроаренов в нейтральной среде арилгидроксиламины синтезируют путем нуклеофильного замещения в активированных аренах.
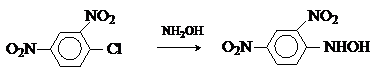
Как
промежуточные продукты восстановления нитроаренов арилгидроксиламины могут быть
окислены в нитрозосоединения (см. выше) и восстановлены в амины путем
каталитического гидирирования или действием металла в кислой среде.
ArNHOH + Zn + 3HCl ® ArNH2 HCl
+ ZnCl2 + H2O
В кислой среде
арилгидроксиламины перегруппировываются аминофенолы, что используется для
получения последних, например:
![]()
Азоксиарены.
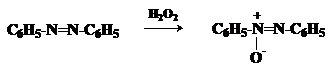
Кроме
описанных выше методов – конденсации нитрозосоединений с арилгидроксиламинами и
восстановления нитроаренов метилатом натрия, азоксиарены могут быть получены окислением
азоаренов пероксисоединениями.
В щелочной среде
азоксиарены восстанавливаются до азо- и далее гидразоаренов.
Образуются при
восстановлении нитроаренов, арилгидразинов и азокси- аренов в щелочной среде,
например:
2ArNO2 +
4Zn + 8NaOH Ar-N=N-Ar + 4Na2ZnO2
+ 4H2O
Несимметричные азосоединения получают конденсацией нитрозосоединений с
аминами (см. выше). Важный метод синтеза азосоединений – реакция азосочетания
будет подробно рассмотрен далее (см. лек.№43)
Азоарены
существуют в виде цис- и транс- изомеров. При облучении более стабильный
транс-изомер превращается в цис-изомер. Обратное превращение происходит
при нагревании.
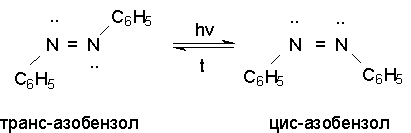
Азосоединения окрашены, многие из них используются в качестве красителей.
Это конечные продукты восстановления нитроаренов в щелочной среде. Гидразоарены
– бесцветные кристаллические вещества, на воздухе окисляются в окрашенные
азосоединения. В препаративных целях окисление проводят действием бромной воды
Ar-NHN-HAr + Br2 + 2NaOH
® Ar-N=N-Ar + 2NaBr + 2H2O
При восстановлении в жестких условиях гидразоарены дают ариламины.
Важным свойством гидразосоединений является перегруппировка в 4,4/-диаминобифенилы.
Это превращение получило название бензидиновой перегруппировки. В
настоящее время этим термином объединяют целую группу родственных
перегруппировок, приводящих к образованию смеси орто- и пара-изомерных
производных диаминобифенила.
При
перегруппировке самого гидразобензола получается смесь диаминов, содержащая 70
% бензидина и 30 % 2,4/-диаминобифенила.
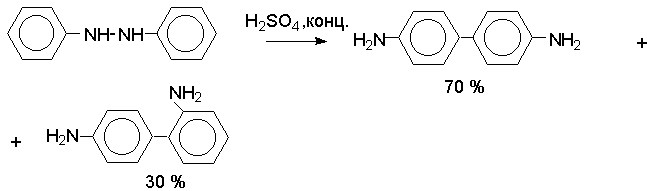
Если пара-положение в одном из бензольных
ядер гидразобензола занято каким-нибудь заместителем, продуктом перегруппировки
оказывается производное дифениламина (так называемая семидиновая
перегруппировка).
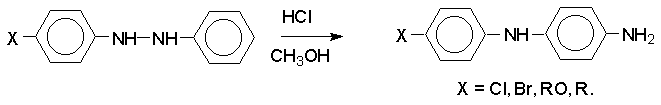
Амины.
Амины можно рассматривать как производные аммиака, в котором атомы водорода
замещены на углеводородные радикалы.
Классификация, изомерия, номенклатура.
В зависимости от числа углеводородных радикалов, связанных с атомом
азота, различают первичные, вторичные и третичные амины, а также четвертичные аммониевые
соли.
RNH2 RR/NH RR/R//N RR/R// R///N+X-
первичные вторичные третичные четвертичные
амины амины
амины аммониевые соли
По типу гибридизации атома углерода, связанного с азотом выделяют следующие
группы аминов.
1) Соединения со связью ![]() .
.
К этой группе относятся алкиламины, а также алкенил- и алкиниламины, в
которых кратная связь удалена от атома азота. Их объединяют под названием алифатические амины. В состав этой
группы входят также циклические амины, содержащие атом азота в цикле, которые
являются гетероциклическими соединениями.
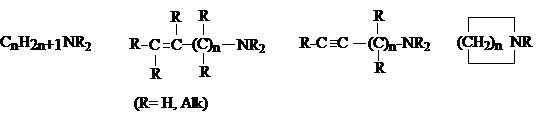
2) Соединения со связью ![]() .
.
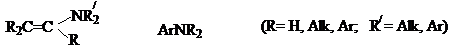
К этой группе принадлежат производные алкенов с атомом азота у атома
углерода, образующего двойную связь – енамины
(виниламины) и амины, содержащие атом
азота, связанный с ароматическим кольцом - ароматические
амины (ариламины).

Названия аминов образуют, добавляя к слову амин названия связанных с
атомом азота углеводородных радикалов.
В другом варианте номенклатуры за основу названия принимают название родоначальной
структуры (самой длинной углеродной цепи, непосредственно связанной с атомом
азота) с добавлением суффикса «амин».
В этом случае вторичные и третичные амины называют как N-замещенные производные первичных аминов.
Если молекула содержит другие функциональные группы, обозначаемые в суффиксе, то аминогруппу обозначают префиксом «амино».
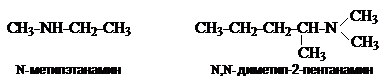
![]()
Названия диаминов образуют от названий соответствующих двухвалентных радикалов
или названия родоначальной структуры с добавлением суффикса «диамин».
Многие ароматические амины имеют тривиальные названия.
Циклические амины называют, используя номенклатуру гетероциклических соединений,
или путем добавления к названию двухвалентного углеводородного радикала
суффикса «имин».
Для аминов характерна изометрия углеродного скелета, изомерия положения
аминогруппы и изомерия между первичными, вторичными и третичными аминами.
1) Алкилирование
аммиака и аминов.
Аммиак взаимодействуют с алкилгалогенидами RX и
другими алкилирующими реагентами (алкилсульфатами, диалкилсульфатами) с
образованием на первой стадии соли алкиламмония, которая в равновесной реакции
с избытком аммиака дает алкиламин. Алкиламин далее вступает в реакцию c алкилгалогенидом с образованием продукта
диалкилирования и т.д. Таким образом последовательно образуются триалкиламин и
соль тетраалкиламмония.
2) Синтез
первичных аминов по Габриэлю.
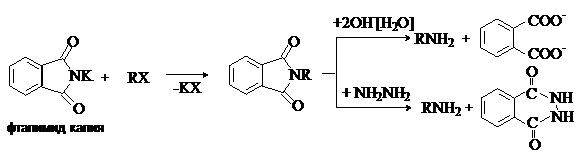
Алкилирование фталимида калия алкилгалогенидами с последующим щелочным
гидролизом или гидразинолизом N-алкилфталимида
позволяет получать первичные амины без примеси вторичных и третичных. Лучше
использовать протекающий в мягких условиях гидразинолиз, приводящий к образованию
не- растворимого в реакционной среде циклического гидразида.
3) Восстановление
азотсодержащих органических соединений.
Нитрилы при восстановлении
дают первичные амины. В промышленности процесс осуществляют путем
каталитического гидрирования.
RCºN + 2Н2 RCH2NH2
В препаративных целях используют восстановление алюмогидридом лития, добавляя
нитрил к избытку восстановителя.
RCºN RCH2NH2
Введение цианогруппы (например, путем нуклеофильного замещения) и её последующее
восстановление – синтетический прием, который позволяет получить амин,
содержащий на один атом С больше, чем исходное соединение.

Амиды карбоновых кислот восстанавливаются до аминов алюмогидридом лития. Из
соответствующих амидов могут быть получены первичные, вторичные и третичные
амины.
Восстановление азотсодержащих производных альдегидов и кетонов – оксимов и гидразонов – дает возможность превращать карбонильные соединения в
первичные амины.
R2C=NOH R2CHNH2
R2C=NNHR R2CHNH2
Для восстановления используют каталитическое гидрирование, комплексные
гидриды металлов (LiAlH4).
Нитросоединения могут быть восстановлены до первичных аминов.
RNO2 RNH2
В качестве восстановителей чаще всего используют металл (Fe, Zn, Sn) и кислоту; алюмогидрид лития. В алифатическом ряду
метод не находит широкого применения из-за ограниченной доступности
алифатических нитросоединений по сравнению с ароматическими.
Восстановление азидов дает
первичные амины.
RN3 RNH2 + N2
Исходные азиды легко могут быть получены из алкилгалогенидов или сульфонатов
путем нуклеофильного замещения.
4) Восстановительное
аминирование карбонильных соединений.
Взаимодействие альдегидов и кетонов с аммиаком в присутствии восстановителя
приводит к первичным аминам.
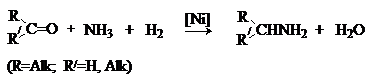
5) Синтез аминов
путем перегруппировок.
Перегруппировка Гофмана:
RCONH2 + Br2
+ 2NaOH ® RNH2 + 2NaBr
+ CO2 + H2O
Перегруппировка Курциуса:
RCON3 RNH2
Реакции подробно рассмотрены ранее (см. лек. №36) В результате образуются
первичные амины без примеси вторичных и третичных. При этом происходит укорочение
углеродной цепи на один атом С.
Физические свойства и строение.
Алифатические амины – бесцветные вещества с неприятным запахом. Низшие амины – жидкости, хорошо растворимые в воде. По растворимости они превосходят спирты с близкой молекулярной массой. Это объясняется образованием между амином и водой водородных связей типа, прочность которых сравнительно велика в силу высокой основности атома азота. Температуры кипения и плавления у третичных аминов ниже, чем у первичных и вторичных с примерно одинаковой молекулярной массой, что связано с ассоциацией последних за счет образования межмолекулярных водородных связей.
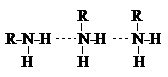
Однако эти межмолекулярные водородные связи слабее, чем у спиртов, по
причине меньшей полярности связи N-Н по сравнению
со связью О-Н. Вследствие этого амины имеют более низкие температуры кипения,
чем спирты с близкой молекулярной массой.
![]()
Амины имеют пирамидальное строение. Величины
углов R-N-R близки к тетраэдрическому – 106-1080.
Считается, что атом азота находится в состоянии sp3-гибридизации,
а четвертым лигандом является неподелённая пара электронов («фантом»-лиганд).
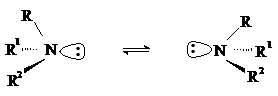
Третичные амины с разными углеводородными радикалами должны быть хиральными,
так как их молекулы не имеют плоскости симметрии. Однако за счет быстрой
пирамидальной инверсии, которая представляет собой акт рацемизации, их
невозможно выделить или зафиксировать в оптически активной форме.
Четвертичные аммониевые соли в случае разных заместителей существуют в
виде пары устойчивых энантиомеров.
Химическое поведение аминов определяется в основном наличием свободной
пары электронов у атома азота, которая обусловливает их основные и нуклеофильные
свойства. Реакции с участием связей N-H и N-C
под действием оснований и нуклеофильных реагентов для аминов менее
характерны.
Основные
свойства.
Алифатические амины являются одними из самых сильных незаряженных оснований
(p![]() ~ 10 - 11). Их водные растворы имеют щелочную реакцию.
~ 10 - 11). Их водные растворы имеют щелочную реакцию.
R3N + H2O
R3NH+ +
OH-
С неорганическими кислотами амины образуют соли, которые в большинстве
случаев хорошо растворимы в воде.
R3N + HX R3N+X-
Основность аминов зависит от их строения и природы растворителя. Сравнение
основности в водных растворах показывает, что алкиламины являются более
сильными основаниями, чем аммиак. Вторичные амины превосходят по основности
первичные. Такой ряд основности согласуется с электронодонорным влиянием
алкильных групп (+I-эффект), которое способствует
делокализации положительного заряда в сопряженной кислоте (ионе аммония) и тем
самым стабилизирует её в большей степени, чем свободный амин. Однако это не объясняет
уменьшения основности при переходе от вторичных аминов к третичным.
|
|
NH3 |
C2H5NH2 |
(C2H5)2NH |
(C2H5)3N |
|
р |
9,25 |
10,80 |
11,09 |
10,85 |
а) Алкилирование
Примеры реакций алкилирования обсуждались при рассмотрении методов получения
аминов.
б) Ацилирование
2RNH2 + R/COX
® R/CONHR
+ RNH3X
2R2NH + R/COX
® R/CONR2
+ R2NH2X
(X=Cl, OCOR/)
Ацилирование аминов функциональными производными карбоновых кислот дает
возможность получать вторичные и третичные амиды из первичных и вторичных
аминов соответственно.
в) Взаимодействие с сульфонилхлоридами
Сульфонилхлориды взаимодействуют с аминами, давая сульфонамиды. Реакция
с бензолсульфонилхлоридом лежит в основе пробы
Гинсберга, позволяющей различать и разделять первичные, вторичные и
третичные амины.
Сульфонамиды, образующиеся из первичных аминов, являются NH-кислотами и со щелочами дают растворимые в воде соли.
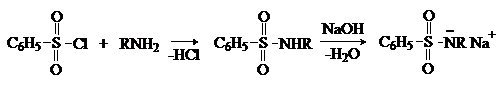
Вторичные амины дают сульфонамиды, которые не содержат кислого водорода
и не растворяются в щелочах.
Третичные амины не реагируют.

г) Нитрозирование
Нитрозирование аминов происходит при взаимодействии с азотистой кислотой
в кислой среде. Неустойчивую азотистую кислоту генерируют действием сильной
кислоты на нитриты. Реакция протекает по-разному в зависимости от типа амина.
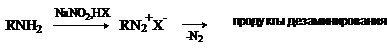
Первичные алифатические амины реагируют с образованием неустойчивых алкилдиазониевых
солей, которые разлагаются с выделением газообразного азота и сложной смеси продуктов дезаминирования.
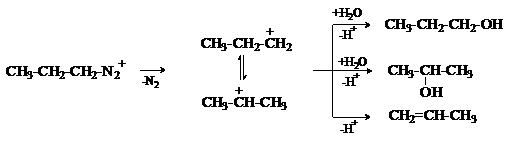
Образование солей диазония – сложный многостадийный процесс, который подробно
будет рассмотрен на примере ароматических аминов.
Разложение катиона алкилдиазония приводит к образованию карбокатиона, который
стабилизируется путем алкилирования присутствующих в реакционной среде
нуклеофилов или путем отщепления протона с образованием алкена. Этим процессам
может предшествовать изомеризация карбокатиона в энергетически более стабильный
ион. Так, разложение катиона н-пропилдиазония
в водном растворе наряду н-пропиловым спиртом дает изопропиловый спирт, а также
продукт элиминирования – пропен.
Со вторичными аминами азотистая кислота образует
нерастворимые в реакционной среде нитрозамины.
R2NH + NaNO2
+ HCl ® R2N-N=O
+ NaCl + H2O
Третичные амины в сильнокислой среде при комнатной температуре с азотистой
кислотой не реагируют.
Нитрозирование аминов препаративного значения не имеет. Аналитическое
значение этих реакций заключается в возможности качественно различить
первичные, вторичные и третичные амины.
д) Галогенирование
Первичные и вторичные амины реагируют с гипогалогенитами с образованием
N-галогенаминов.
RNH2 RNHX RNX2
R2NH R2NX
(X= Cl, Br)
N-галогенамины – сильные окислители и галогенирующие
реагенты.
Амины дают разные продукты окисления, состав которых зависит от природы
окислителя и строения амина.
Перекись водорода и надкислоты окисляют третичные амины до N-оксидов.
R3N + HOOH ® R3N+-O-
+ H2O
В случае первичных и вторичных аминов первоначально образующиеся N-оксиды перегруппировываются в производные гидроксиламина.
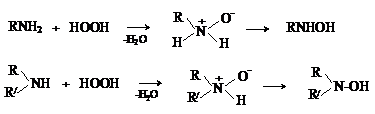
Такое окисление протекает сложно, так как гидроксиламины сами легко
окисляются. В случае первичных аминов конечными продуктами окисления
являются нитросоединения, например:
CH3(CH2)5NH2
CH3(CH2)5NO2
Первичные амины, в которых аминогруппа соединена с третичным атомом углерода,
окисляются в нитросоединения перманганатом калия в водном ацетоне.
R3CNH2 R3CNO2
Амины, содержащие атомы водорода в a-положении, при действии сильных окислителей (KMnO4) дают
смесь веществ, в которой преобладают карбонильные соединения. Процесс протекает
через промежуточное образование иминов, которые при гидролизе дают альдегиды
или кетоны.
RR/CHNH2
RR/C=NH
RR/CO
(R=Alk; R/=H, Alk)
Кислотные свойства.

Первичные и вторичные алифатические амины являются очень слабыми NH-кислотами (pKа~33-35). Их кислотные свойства проявляются при действии
щелочных металлов или таких сильных основания, как металлоорганические соединения.
Образующиеся алкил- и диалкиламиды металлов – очень сильные основания.
Диалкиламиды, содержащие вторичные или третичные алкильные радикалы (например,
диизопропиламид лития), представляют интерес для органического синтеза как
ненуклеофильные основания. Будучи сильными основаниями, они обладают низкой
нуклеофильностью по причине стерических затруднений, возникающих при атаке
электрофильных центров за исключением протона. Их используют в органическом
синтезе для отрыва протона и генерирования карбанионов.
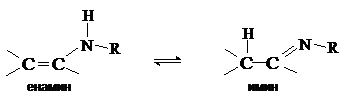
Енамины (виниламины) устойчивы в том случае, если при атоме азота нет
атомов водорода. Такие енамины можно рассматривать как азотистые аналоги виниловых
эфиров. В противном случае енамины нестабильны и перегруппировываются в имины,
подобно тому, как енолы изомеризуются в карбонильные соединения.
Основной метод получения енаминов – взаимодействие карбонильных соединений со вторичными аминами в присутствии кислотных катализаторов и средств, связывающих воду.
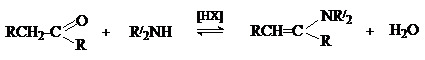
Реакция протекает по общему для присоединения азотистых оснований к карбонильной группе механизму.
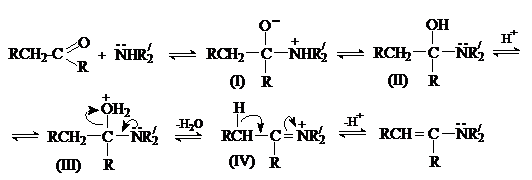
Отщепление воды от интермедиата (III) приводит к образованию иммониевого
иона (IV), который при отсутствии водорода у атома азота стабилизируется путем
отщепления протона от b-углеродного атома.
Молекула енамина представляет собой р-p-сопряженную систему, строение которой можно отразить набором двух резонансных структур.
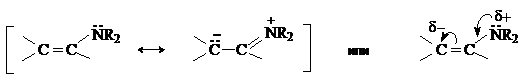
Таким образом, молекула енамина содержит два нуклеофильных центра –
атом азота и атом углерода в b-положении, который несет частичный отрицательный заряд.
а) Протонирование и гидролиз
Енамины являются слабыми основаниями. Их протонирование может протекать
как по атому азота, так и по атому углерода.
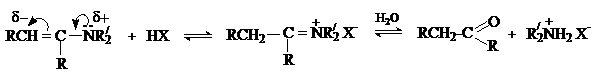
Образующаяся при протонировании по b-углеродному атому соль иммония гидролизуется, давая
исходное карбонильное соединение и вторичный амин.
Гидролиз енаминов – процесс, обратный их образованию, и протекает по
такому же механизму.
б) Алкилирование
Алкилирование енаминов алкилгалогенидами и другими алкилирующими реагентами протекает, как правило, по b-углеродному атому. Последующий гидролиз иммониевой соли приводит к карбонильному соединению.
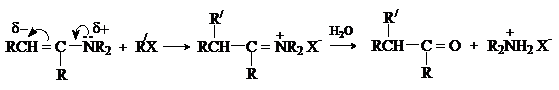
Последовательность превращений – получение енамина из карбонильного соединения,
алкилирование, гидролиз приводит к алкилированию исходного карбонильного
соединения по a-положению и носит название реакция
Сторка. Этот метод имеет преимущества перед алкилированием кетонов, так как
требует более мягких условий и дает в основном продукты моноалкилирования.
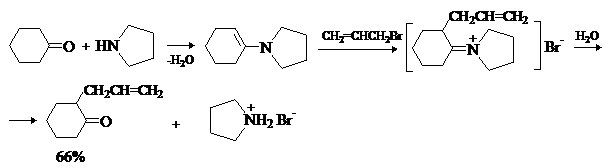
Для проведения этой реакции чаще всего используют циклические амины –
пирролидин, пиперидин, морфолин. Лучшие результаты достигаются при использовании
активных галогенидов – аллил- и бензилгалогенидов, a-галогензамещенных производных простых и сложных
эфиров. Например:
в) Ацилирование
При действии галогенангидридов и ангидридов кислот енамины дают продукты С-ацилирования. Последующий гидролиз приводит к дикарбонильному соединению.
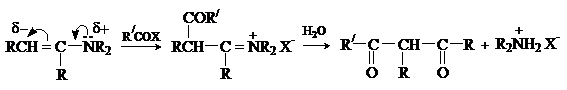
1)
Восстановление ароматических нитросоединений
ArNO2 ArNH2
Для восстановления в препаративных целях используют металл (Fe, Zn, Sn)
и кислоту, соли металлов в низших степенях окисления (SnCl2, TiCl3),
сульфиды щелочных металлов, в промышленности применяют в основном каталитическое
гидрирование.
2)
Алкилирование
Реакция аналогична алкилированию алифатических аминов. В качестве алкилирующих
реагентов используют алкилгалогениды, алкилсульфаты, спирты.
3)
Арилирование
Галогенарены реагируют с аммиаком и аминами в жестких условиях. Процесс
катализируется медью и ее соединениями.
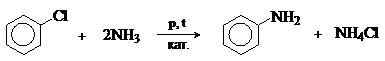
Реакция замещения галогена протекает легче при наличии в орто- и пара-положениях электроноакцепторных групп (NO2, CN).
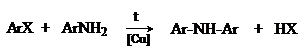
Галогенарены взаимодействуют с ариламинами в присутствии меди с образованием
диариламинов (реакция Ульмана).
Физические
свойства и строение.
Ароматические амины – бесцветные жидкости или твердые вещества. При хранении
быстро темнеют вследствие окисления кислородом воздуха.
Аминогруппа и ароматическое кольцо образуют сопряженную систему. Аминогруппа
проявляет электронодонорные свойства за счет +М-эффекта.
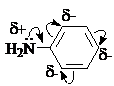
Ароматические амины обладают сильными электронодонорными свойствами, на
что указывают низкие энергии ионизации (для анилина 7,7 эВ, для фенола 8,4 эВ).
Для ариламинов характерны реакции с электрофильными реагентами. Местом
атаки электрофила может быть атом азота или ароматическое кольцо.
Основные
свойства.
Ароматические амины обладают меньшей основностью, чем алифатические
амины и аммиак (p![]() ~ 3 – 5). Причина низкой основности ариламинов – стабилизация
свободного амина за счет делокализации неподеленной пары электронов азота по
ароматическому кольцу и потеря энергии
стабилизации при нарушении сопряженной системы в результате протонирования.
~ 3 – 5). Причина низкой основности ариламинов – стабилизация
свободного амина за счет делокализации неподеленной пары электронов азота по
ароматическому кольцу и потеря энергии
стабилизации при нарушении сопряженной системы в результате протонирования.
Дифениламин и трифениламин имеют еще большие возможности для делокализации
пары электронов азота, что приводит значительному снижению основности.
Трифениламин практически не обладает основными свойствами.
Введение заместителей в ароматическое кольцо влияет на основность амина.
Влияние заместителей проявляется в соответствии с их электронными эффектами.
|
|
p |
|
p |
|
п-CH3C6H4NH2 |
5,1 |
п-O2NC6H4NH2 |
1,0 |
|
C6H5NH2 |
4,6 |
(C6H5)2NH |
0,78 |
|
м-С2NC6H4NH2 |
2,5 |
|
|
Реакции
с С-электрофилами.
Важнейшими реакциями этого типа являются алкилирование и ацилирование,
которые протекают по атому азота и аналогичны реакциям алифатических аминов.
Ароматические амины менее реакционноспособны из-за меньшей основности атома
азота.
Реакции
ароматического электрофильного замещения.
а) Галогенирование
Аминогруппа является сильным активирующим заместителем
и ориентантом I рода. Галогенирование свободных
аминов протекает очень легко и часто приводит к полигалоидпроизводным.
Например, анилин при действии бромной воды мгновенно превращаются в нерастворимое
2,4,6-трибромпроизводное.
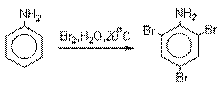
Для получения моногалогенпроизводных активирующее действие аминогруппы
снижают путем ацилирования. После снятия ацильной защиты путем гидролиза
получают свободный амин.
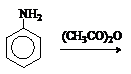
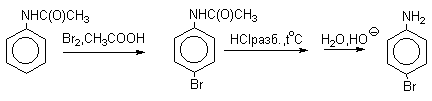
б) Нитрование
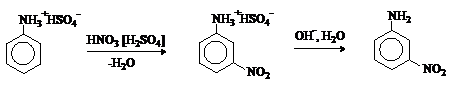
При нитровании нитрующей смесью амины окисляются. Кроме того, из-за солеобразования
по аминогруппе образуется м-изомер (-NH3+ -
ориентант II рода).
Для введения нитрогруппы в орто-
или пара-положение к аминогруппе последнюю
защищают ацилированием. Варьируя условия реакций (температуру, нитрующие
агенты), можно проводить нитрование региоселективно.
После снятия ацетильной защиты получают свободные орто- и пара-нитроанилины.
в) Сульфирование
Сульфированием ароматических аминов получают
аминосульфокислоты. В 90-100%-ной серной кислоте или олеуме амин полностью
находится в протонированной форме. Аммониевая группа NH3+
как сильный электроакцепторный заместитель вызывает резкое замедление
реакции сульфирования и ориентирует замещение в мета-положение.
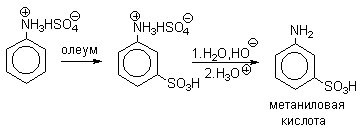
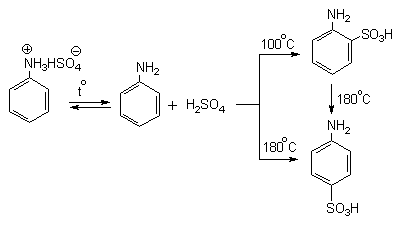
Для получения орто-
и пара-аминобензолсульфокислот
используют «метод запекания». Процесс осуществляют при длительном нагревании
гидросульфатов ароматических аминов при 100-200оС в сухом виде или в
высококипящих растворителях. При температуре около 100оС образуется
практически чистый орто-изомер
(ортаниловая кислота, продукт кинетического контроля), а при 180-200оС
- пара-изомер (сульфаниловая кислота,
продукт термодинамического контроля).
Диазосоединения.
Диазосоединения содержат группу N2, причем
один атом азота связан с органическим радикалом, а другой - с гетероатомом.
Ароматические и алифатические диазосоединения сильно различаются по строению,
стабильности и реакционной способности.
Ароматические диазосоединения.
Классификация и номенклатура.
Ароматические диазосоединения имеют общую формулу ArN2X. В зависимости от типа связи между фрагментами ArN2 и X
различают следующие типы диазосоединений.
Соединения, содержащие ионную связь (соли диазония): ArN2+X-
X- – анион сильной кислоты с низкой нуклеофильностью, в том числе комплексный ион (Cl-, Br- HSO4-, BF4-, SbF6-, PF6-), например:
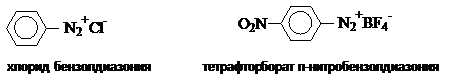
Соединения, содержащие ковалентную связь:
X= -CN, -OCOCH3,
-I, -SO3H, -OH, -OAr, -NH-Ar
Например:
|
C6H5-N=N-CN |
C6H5-N=N-OH |
C6H5-N=N-O-
Na+ |
|
Бензолдиазоцианид |
бензолдиазогидроксид |
бензолдиазотат натрия |
Основной тип ароматический диазосоединений – соли
арендиазония.
Методы получения солей арендиазония
Основной метод получения солей арендиазония – диазотирование. Процесс диазотирования
выражается следующим суммарным уравнением.
ArNH2
+ NaNO2 + 2 HX ®
ArN+=N X - + NaX + 2 H2O
(X= Cl, HSO4, BF4 и др.)
Условия проведения реакции диазотирования:
Диазотирование – экзотермическая реакция,
сопровождающаяся выделением большого количества теплоты, поэтому процесс
проводят при охлаждении, поддерживая температуру в интервале 0 – 5о,
чтобы не допустить разложения термически неустойчивой соли арендиазония.
Диазотирование осуществляется четырьмя основными
методами, выбор каждого из которых определяется природой исходного амина.
1. Метод
"прямого" диазотирования применим для диазотирования анилина, алкил-,
галоген - и алкоксианилинов. Раствор одного эквивалента нитрита натрия при
охлаждении до 0 - 5о постепенно добавляют к водному раствору одного
эквивалента амина с тремя эквивалентами сильной минеральной кислоты.
2.
"Обратный" метод диазотирования используют для плохо растворимых в кислотах
аминов, например, сульфаниловой и антраниловой кислот, для которых характерно
образование внутренних солей. В этом случае щелочной раствор диазотируемого
амина и нитрита натрия постепенно вводят в водный раствор охлажденной до +5о
разбавленной минеральной кислоты.
3. Для
диазотирования аминофенолов, которые легко окисляются азотистой кислотой,
реакцию проводят с добавлением солей меди, препятствующих окислению амина.
4. Для
диазотирования слабоосновных аминов, содержащих одну или несколько нитрогрупп,
три- или тетрагалогенанилинов (Hal=Cl,Br), амин растворяют в смеси
концентрированных H2SO4 - H3PO4 или
H2SO4 - CH3COOH и полученный раствор при
охлаждении прибавляют к нитрозилсерной кислоте.
Полученный одним
из описанных выше методов водный раствор соли диазония сразу используют для
дальнейших превращений. Предварительно следует убедиться в полноте протекания
диазотирования, т.е. проверить отсутствие в реакционной массе непрореагировавших
исходных веществ. При добавлении к порции диазораствора ацетата натрия,
снижающего кислотность среды, при наличии исходного амина образуется триазен –
нерастворимый в воде оранжевый осадок.
![]()
Азотистую
кислоту обнаруживают с помощью иодкрахмальной бумаги. Избыток азотистой кислоты
удаляют добавлением мочевины до прекращения выделения газообразных продуктов из
реакционной смеси.
Физические
свойства и строение.
Соли арендиазония – бесцветные кристаллические вещества. Хлориды, гидросульфаты
и нитраты хорошо растворимы в воде и используются в основном в виде водных
растворов. В твердом виде они неустойчивы и взрываются от трения и от
нагревания. Соли арендиазония с некоторыми комплексными анионами (PF6-,
SbF6-, BF4-) нерастворимы в воде и
достаточно стабильны при хранении их в сухом виде в течение длительного
времени.
В целом соли арендиазония гораздо устойчивее алифатических аналогов,
что обусловлено стабилизирующим влиянием p-системы ароматического ядра, которое участвует в
делокализации положительного заряда.
Электронодонорные заместители в о-
и п-положениях обеспечивают дополнительную
стабилизацию арендиазониевого катиона.
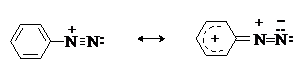
Сама диазониевая группа может быть представлена набором из двух резонансных структур.

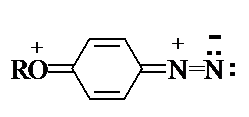
Наибольший вклад в резонанс вносит структура (I). По данным рентгеноструктурного
анализа связь азот-азот по длине соответствует тройной (0,11 нм). ИК-спектры
содержат полосу поглощения в области 2100-2300 см-1, что соответствует
валентным колебания связи NºN. Второй атом азота несет частичный положительный
заряд (структура (II)) и является электрофильным центром в реакциях диазониевых
солей. Диазониевая группа является сильнейшим электроноакцептором и превосходит
в этом отношении нитрогруппу.
1. Равновесия между различными формами
диазосоединений в водных растворах. Образование и строение диазотатов.
Форма
существования диазосоединений в растворе зависит от рН. Катионы диазония могут
существовать в кислой среде и в средах, близких к нейтральной. При обработке
растворов солей диазония щелочью образуются диазотаты.
ArN2+X- + 2NaOH ® Ar-N=N-O- Na+ + NaX + H2O
2. Реакции солей арендиазония.
Все реакции солей диазония можно разделить на четыре
большие группы, различающиеся по механизму взаимодействия катиона диазония с
нуклеофильным агентом Х-.
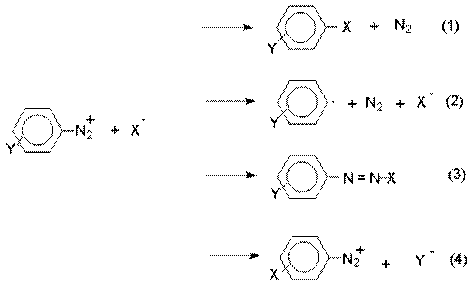
Реакции типа (1) и (4) - нуклеофильное ароматическое
замещение. В реакциях группы (1) имеет место замещение самой диазогруппы. В
реакциях типа (4) диазогруппа, как очень сильный акцептор, активирует
нуклеофильное замещение группы Y в орто-
или пара-положении к диазогруппе. В
реакциях группы (2) происходит радикальный распад соли диазония. Образующийся
при этом арильный радикал Ar претерпевает затем дальнейшее превращение. Третий тип
реакций предполагает атаку нуклеофильным агентом крайнего атома азота
диазониевого катиона.
По формальному признаку реакции солей диазония можно
разделить на две большие группы: реакции идущие с выделением (1,2) и без
выделения (3,4) азота.
Реакции, идущие с выделением азота.
Замещение диазогруппы – один из основных методов
введения заместителей в ароматическое кольцо. Замещение протекает как по гетеролитическому, так и по
гомолитическому механизму. Характер разрыва связи углерод-азот в катионе
арендиазония зависит прежде всего от природы нуклеофильного агента. Гетеролиз
связи С-N имеет место при взаимодействии с жесткими нуклеофильными агентам –
водой, фторид-ионом и при проведении реакции в растворителях с электрофильными
или малонуклеофильными свойствами. Гомолитическому расщеплению связи
углерод-азот благоприятствует рост нуклеофильных свойств растворителя и
использование мягких нуклеофильных агентов (J- , SCN-).
Все эти анионы относятся к сильным восстановителям, что облегчает перенос
одного электрона от реагента к катиону диазония. Эту же функцию в отдельных
случаях может выполнять и растворитель.
Для многих реакций переход от гетеролитического к
гомолитическому механизму происходит очень легко при введении в раствор солей
одновалентной меди.
Замещение диазогруппы на гидроксил.
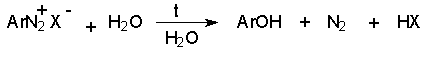
Замещение диазогруппы на фтор (реакция Шимана).
Замещение диазогруппы на фтор происходит при
термическом разложении тетрафторборатов арендиазония.
ArN2+BF4-
ArF + N2
+ BF3
Реакция протекает, вероятно, через промежуточное образование арилкатиона.
Выходы арилфторидов сильно колеблются в зависимости от природы заместителя в
бензольном кольце арендиазокатиона. Увеличению выхода способствует наличие в
бензольном кольце электронодонорных
заместителей. Реакция Шимана – один из основных методов введения фтора в
ароматическое кольцо.
Замещение диазогруппы на иод.
ArN2+X-
+ KI ® ArI + N2
+ KX
Замещение диазогруппы на йод осуществляется по
принципиально иному механизму и представляет собой ион-радикальный процесс, в
котором иодид-ион выступает в роли одноэлектронного восстановителя.
Замещение диазогруппы на хлор-, бром-, циан- и
нитрогруппу (реакция Зандмейера).
Выходы
арилхлоридов, арилбромидов и арилцианидов в реакциях солей арендиазония с
хлорид-, бромид- и цианид-ионами в отсутствие катализаторов невелики и редко
превышают 20%. Для получения этих соединений используют реакцию Т. Зандмейера,
который обнаружил, что такое замещение эффективно катализируется солями меди
(I). Для введения хлора и брома используют раствор галогенида меди (I) в
соответствующей галогеноводородной кислоте.
ArN2+Cl- ArCl + N2
ArN2+Br- ArBr + N2
Для введения
цианогруппы нейтральный раствор соли диазония смешивают с раствором
комплексного цианида.
Замещение диазогруппы на водород (дезаминирование
первичных ароматических аминов).
Замещение
диазогруппы на водород происходит при действии восстановителей. В качестве
восстановителей используют спирты.
ArN2+X- + RCH2OH
® ArH + R-CH=O + N2
+ HX
Реакция
протекает по цепному радикальному механизму.
Реакции, идущие без выделения азота.
Реакция азосочетания.
Соли диазония
реагируют с фенолами и ароматическими аминами с образованием ярко окрашенных
азосоединений.
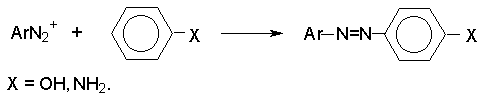
Азосочетание
относится к типичным реакциям электрофильного ароматического замещения,
скорость которых зависит от электрофильных свойств катиона диазония (диазосоставляющей)
и от электронодонорных свойств ArX (азосоставляющей).
Восстановление солей арендиазония в арилгидразины.
Существует два общих метода восстановления солей
диазония до арилгидразинов.
Первый способ заключается в восстановлении катиона
арендиазония смесью сульфита и гидросульфита натрия. На первой стадии образуется
ковалентный диазосульфонат, который далее восстанавливается до соли арилгидразин-N–N/-дисульфокислоты,
гидролизующейся далее до арилгидразина.
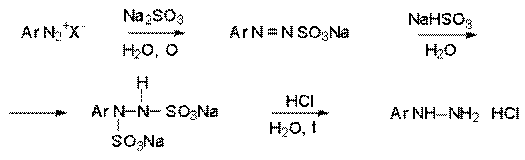
Другой способ состоит в восстановлении солей диазония двухлористым
оловом в соляной кислоте.
![]()
Алифатические диазосоединения.
Соли алкандиазония крайне нестабильны. Относительно
устойчивыми диазосоединениями алифатического ряда являются диазоалканы, которые
можно рассматривать как внутренние соли алкандиазония.
CH2N2
(CH3)2CN2
диазометан
2-диазопропан
Строение и физические свойства.
Диазоалканы – газообразные или жидкие вещества желтого
цвета, взрывоопасные и ядовитые. Газообразный диазометан разлагается со взрывом
при комнатной температуре при соприкосновении со шлифованной или шероховатой
поверхностью, поэтому его используют и хранят в виде раствора в эфире при низких температурах.
Диазоалканы имеют структуру внутренних солей, которая может быть представлена набором резонансных структур.
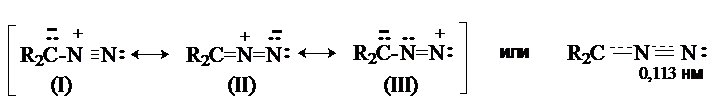
Длина связи N-N близка к тройной, т.е. структура диазоалканов близка
к структуре биполярного иона (I).
Производные диазоалканов с электроноакцепторными
группами - a-диазоэфиры, a-диазокарбонильные соединения более стабильны за счет
делокализации заряда с участием карбонильной группы.
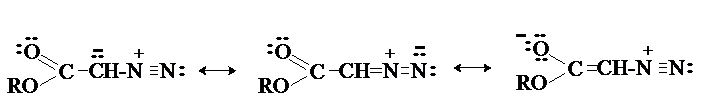
Методы получения.
Диазоалканы не могут быть получены нитрозированием первичных аминов, так как образующиеся алкандиазониевые соли спонтанно разлагаются с выделением азота (см. лек.№42). Диазометан получают обработкой N-нитрозометилмочевины или N-нитрозометилуретана щелочью.

При взаимодействии a-аминоэфиров с азотистой кислотой дезаминирования не происходит. Из a-положения, активированного карбонильной группой,
отщепляется протон и образуются a-диазоэфиры.
Нитрозированием a-аминокетонов
получают a-диазокетоны.
Другой способ получения a-диазокетонов – ацилирование
диазометана ацилгалогенидами.
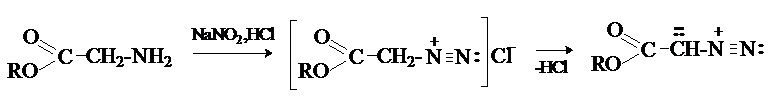
1. Реакции с протонными кислотами.
Диазоалканы являются С-основаниями и легко реагируют с соединениями, содержащими кислый водород. Реакция протекает через стадию протонирования диазоалкана с образованием нестабильного катиона алкандиазония, который реагирует с нуклеофилом с выделением азота.
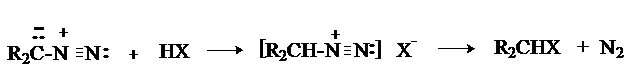
Таким образом
диазометан количественно реагируют с гидроксильной группой карбоновых кислот и
фенолoв. Обладающие меньшей кислотностью спирты реагируют при катализе эфиратом
трехфтористого бора.
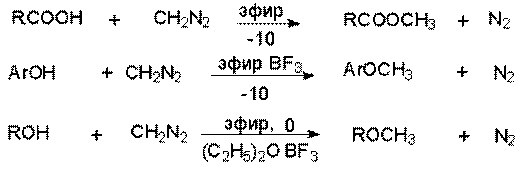
a-Диазоэфиры реагируют аналогично, но в силу меньшей
основности менее реакционноспособны.
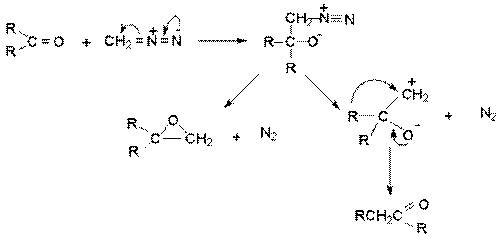
2. Реакции с карбонильными соединениями.
Диазоалаканы как нуклеофилы присоединяются по карбонильной группе альдегидов
и кетонов. Менее реакционноспособные a-диазоэфиры реагируют только с альдегидами. Дальнейшие
превращения приводят в случае альдегидов к метилкетонам, из кетонов образуются
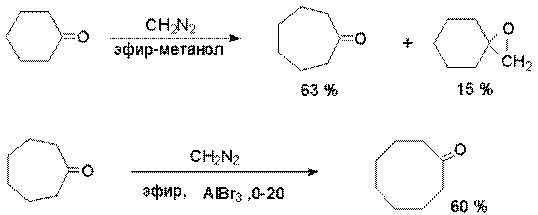
их гомологи. В обоих случаях побочно образуются оксираны.
Реакция имеет препаративное значение для синтеза некоторых циклических
кетонов как метод расширения цикла.
Взаимодействие диазометана с хлорангидридами приводит к a-диазокетонам.
3. Реакции биполярного присоединения.
Диазоалканы
реагируют с активированными алкенами с образованием пиразолинов, а с ацетиленом
дают пиразолы. При повышенных температурах пиразолины отщепляют азот и
превращаются в производные циклопропана.
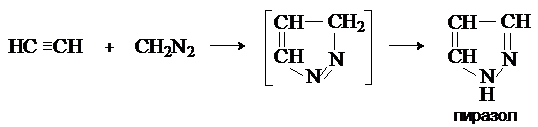
4. Фотохимическое и термическое разложение.
При облучении
или нагревании диазоалканы разлагаются с образованием карбенов. Диазометан при
облучении разлагается на карбен - метилен и азот.

Карбены –
высокореакционноспособные частицы, которые претерпевают дальнейшие превращения,
например, присоединяются к двойной связи алкенов с образованием производных
циклопропана. Разновидностью этого способа генерации карбена является каталитическое
разложение диазосоединений, и в первую очередь, диазоуксусного эфира в
присутствии солей меди, а также ионов других переходных металлов.