Лекция
№23
РЕАКЦИИ НУКЛЕОФИЛЬНОГО ОТЩЕПЛЕНИЯ
План
- Реакции
нуклеофильного отщепления.
- Механизм
реакций отщепления. Направление отщепления.
- Конкуренция
реакций нуклеофильного замещения и отщепления и роль изомеризации.
Эти реакции называют еще реакциями элиминирования (англ.
elimination). Их можно классифицировать по взаимному расположению отщепляемых
групп (1,1-, 1,2-, 1,3-элиминирование и т.д.).
Наибольшее практическое значение имеют реакции 1,2-отщепления, ведущие к образованию ненасыщенных веществ (процессы дегидрогалогенирования, дегидратации и др.):
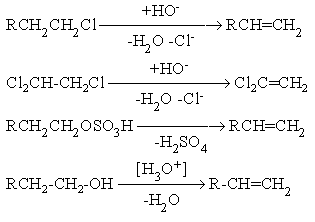
а также отдельные реакции 1,3 и 1,4-отщепления с образованием гетероциклических соединений
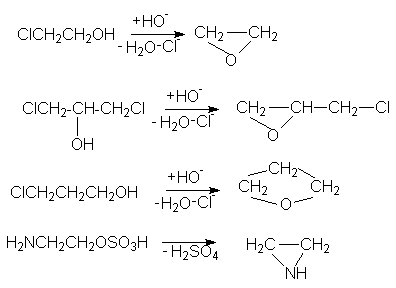
Механизм реакций отщепления
В зависимости от условий реакции и строения реагирующих веществ 1,2-отщепление может протекать по разным механизмам. По очередности разрыва связей С–Х и С–Н они подразделяются на механизмы Е1, Е2 и Е1св, где цифры 1 и 2 – показывают молекулярность скорость-определяющей стадии, индекс “св” означает сопряженное основание (англ. conjugated basе).
Реакция Е1 обычно конкурирует с SN1 и имеет с ним общую скорость-определяющую стадию гетеролиза связи С-Х
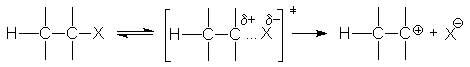
Образовавшийся карбкатион взаимодействует далее с каким-либо основанием или растворителем, выполняющим функцию основания, с отщеплением протона и образованием алкена:
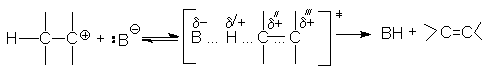
В соответствии с характером лимитирующей стадии протеканию Е1 благоприятствуют те же факторы, что и реакции SN1, а скорость реакции выражается кинетическим уравнением
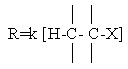
подобным уравнению скорости SN1 – реакции.
Как указывалось выше, к этим факторам относятся разветвление алкильной группы и наличие электронодонорных заместителей при реакционном центре субстрата, уменьшение энергии связи С–Х, наличие растворителя и катализаторов, способных к электрофильному взаимодействию с Х. Типичный пример представляет дегидратация спиртов, катализируемая протонными или апротонными кислотами, которые стабилизируя уходящую группу, способствуют ее эффективному отщеплению и образованию карбкатиона:
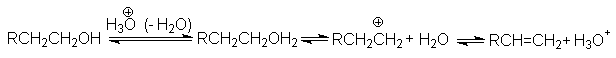
Таким образом, протонированный спирт претерпевает мономолекулярное расщепление, определяя Е1 – характер реакции.
Поскольку стабильность ионов карбония возрастает в ряду
трет. R+ > втор. R+ > перв. R+
то дегидратация наиболее легко протекает у третичных спиртов, в то время как для первичных спиртов требуются наиболее жесткие условия.
Если в субстрате отсутствуют структурные возможности для стабилизации карбкатиона, а реагент представляет собой сильное основание, то реакция отщепления протекает по механизму Е2. В этом случае разрыв связей С-Н и С-Х происходит синхронно в процессе бимолекулярной элементарной реакции
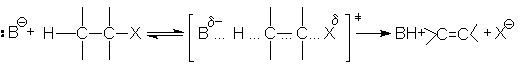
и скорость такой реакции описывается кинетическим уравнением второго порядка
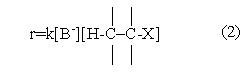
подобным уравнению для реакции SN2.
По этому механизму протекают реакции расщепления н-хлоралканов, эфиров серной кислоты и др.
Реакция Е2 ускоряется электроноакцепторными группами, повышающими кислотность отщепляемого атома водорода. К таким группам относятся СF3, NO2, SO2, CN, C=O, наличие которых при том же углеродном атоме, что и отщепляемый водород, существенно ускоряет реакцию. Примером может служить катализируемая основаниями дегидратация b -кетоспиртов
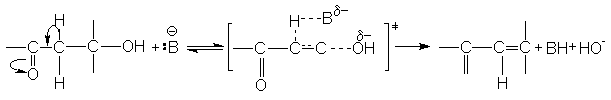
![]()
Если протон в субстрате обладает достаточно высокой кислотностью, а группа Х отщепляется трудно, реакция протекает по механизму Е1св, когда происходит предварительное отщепление протона по быстрой равновесной реакции
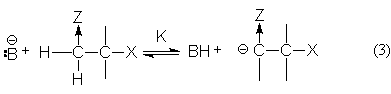
а группа Х отщепляется в следующей медленной мономолекулярной стадии распада карбкатиона
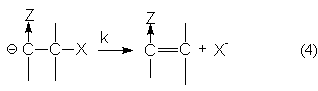
Роль электроноакцепторной группы Z заключается в поляризации отщепляемого протона и стабилизации промежуточного аниона.
Скорость реакции в соответствии с характером лимитирующей стадии выражается уравнением
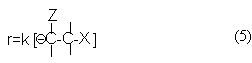
Концентрация карбаниона определяется равновесием (3)
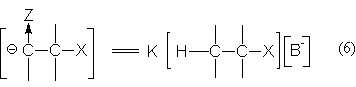
Подставляя выражение (6) в уравнение (5) приходим к кинетическому уравнению реакции Е1св
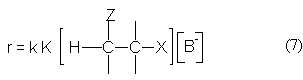
Из уравнений (2) и (7) видно, что механизмы Е2 и Е1св кинетически неразличимы. Различие между ними можно выявить на основе анализа активационных параметров брутто-реакций. Так, константа скорости реакции SN2 как константа истинной бимолекулярной реакции будет характеризоваться отрицательными энтропиями активации, тогда как брутто-константа реакции Е1св должна характеризоваться положительной энтропией активации.
Примером Е1св-реакции является 1,3-отщепление НСl от хлоргидринов с образованием оксиранов
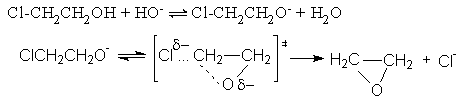
Направление отщепления
Если атом углерода, несущий отщепляемую группу Х, имеет два или три соседних углеродных атома, содержащих С-Н–связи, возможно образование нескольких изомерных алкенов. Направление отщепления в этом случае определяется правилом Зайцева, согласно которому водород предпочтительно отщепляется от наименее гидрированного атома углерода с образованием разветвленного алкена. Например, при щелочном дегидробромировании 3-бром-2,3-диметилпентана доминирующим продуктом реакции будет 2,3-диметилпентен-2
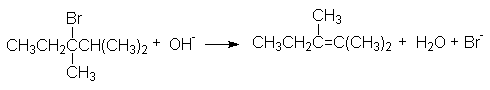
В основе такого направления отщепления лежит более высокая термодинамическая стабильность разветвленных алкенов.
Правило Зайцева применимо к реакциям, протекающим и по Е1 и по Е2-механизмам.
В несимметричных дигалогенэтанах направление отщепления также соответствует правилу Зайцева. Однако в этом случае в основе его лежит более высокая кислотность атомов водорода при наименее гидрированном, углеродном атоме из-за наличия у него большего числа электрноакцепторных заместителей
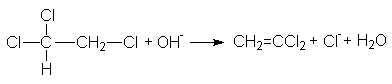
При разложении четвертичных аммониевых и сульфониевых соединений направление отщепления противоположное и определяется правилом Гофмана, согласно которому водород отщепляется от наиболее гидрированного атома углерода, в результате чего образуется менее разветвленный алкен:
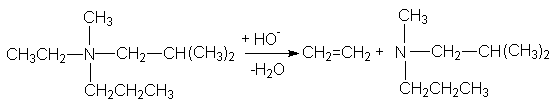
Такой порядок расщепления связан с близостью
механизмов этих реакций к механизму Е1св. В переходном состоянии
записанной реакции связь С-Н ослаблена сильнее, чем связь С-N, и промежуточный
карбанион (или близкое к нему по структуре переходное состояние) оказывается
менее дестабилизированным алкильными группами при образовании менее
разветвленного алкена.
Конкуренция реакций нуклеофильного замещения и отщепления и роль изомеризации
Если структура субстрата делает возможным его расщепление, эта реакция всегда конкурирует с замещением. Очевидно, что состав продуктов будет зависеть от соотношения скоростей этих конкурирующих реакций, определяемых различными факторами. При конкуренции SN1– и Е1-реакций
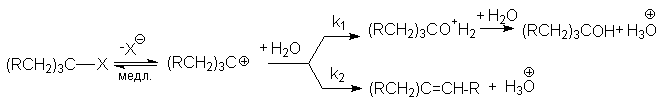
cостав продуктов не зависит от общей скорости и определяется соотношением констант скоростей быстрых стадий k1/k2. На состав продуктов не влияют ни способ образования карбкатиона, ни природа отщепляемой группы Х, что может служить подтверждением SN1– и Е1-механизма.
Удлинение и разветвление алкильной группы в исходном соединении приводит к росту доли алкена в продуктах реакции при конкуренции SN1– и Е1-механизмов. Это объясняется большей стабилизацией переходного состояния образования более разветвленного алкена из-за его высокой термодинамической стабильности. переход к более сольватирующей среде способствует увеличению удельного веса продукта замещения, так как переходное состояние быстрой стадии реакции SN1 характеризуется большей степенью локализации заряда
(RCH2)3Cd+… Od + H2
чем переходное состояние конкурирующей с ней реакции отщепления
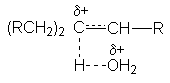
Благодаря этому первое сольватируется сильнее, что приводит к большему понижению энергетического барьера реакции замещения.
При конкуренции реакций SN2 и Е2
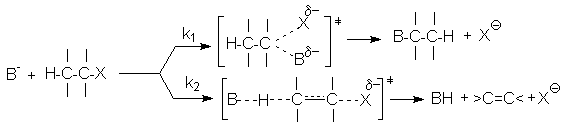
удлинение и разветвление алкильного радикала субстрата приводит к увеличению доли продукта отщепления, как и в случае конкуренции SN1–E1. В основе такого влияния лежат две причины: более высокая термодинамическая стабильность разветвленных алкенов и возрастание стерических препятствий в реакции SN2 с увеличением размера алкильной группы субстрата. В то же время в реакции отщепления Е2 атака основания направлена на периферийный атом водорода и практически не имеет стерических препятствий. Из приведенных соображений следует, что при переводе механизма из SN1– Е1 в SN2– Е2 должен повышаться выход продукта отщепления.
В реакции SN2 по сравнению с Е2 переходное состояние характеризуется большей поляризацией отрицательного заряда, поскольку в случае SN2 заряд распределен по трем атомам, а в случае Е2 – по пяти. Поэтому при переходе к более сольватирующему растворителю энергия переходного состояния SN2 будет понижаться в большей степени, чем энергия переходного состояния Е2, что приводит к увеличению доли продукта замещения. Наоборот, при использовании неполярных апротонных растворителей увеличивается выход алкена.
Важным фактором регулирования состава продуктов замещения-отщепления является температура. Как правило, энергия активации реакции отщепления (Е2) выше, чем энергия активации замещения (SN2). Поэтому увеличение температуры приводит к увеличению удельного веса продукта отщепления. Понижение температуры приводит к противоположному результату. В случае конкуренции SN1– Е1 температура в меньшей степени влияет на состав продуктов, так как различия в энергиях активации конкурирующих реакций из-за их экзотермичности существенно ниже.
Большое значение в конкуренции реакций замещения и отщепления имеет природа нуклеофильного реагента. Успех реакций SN2 определяется его сродством к углеродному атому, имеющему частичный положительный заряд, т.е. нуклеофильностью. При Е2- и Е1св-отщеплении нуклеофил атакует положительно заряженный атом водорода и его активность определяется его основностью. Различие между нуклеофильностью и основностью часто является очень большим и это во многом определяет направление реакции. Для таких сильных оснований, как едкие щелочи и третичные амины наиболее характерны реакции отщепления, что обусловливает их практическое применение для процессов дегидрохлорирование. Наоборот, для относительно сильных нуклеофилов, но слабых оснований (HS–, RS–, ArO–, NH3, Br–, J–) преобладающим направлением является замещение. Этим пользуются при промышленном гидролизе хлорпроизводных в спирты, когда применение едких щелочей дает слишком большой выход алкенов. Замена щелочей на более слабое основание (карбонат натрия, его буферные смеси со щелочью или бикарбонат натрия) позволяет существенно повысить выход спиртов.
В случае конкуренции SN1– Е1, различия в нуклеофильности и основности не играют такой существенной роли по двум причинам: во-первых, нуклеофильная атака центрального атома углерода карбкатиона не встречает серьезных стерических препятствий из-за плоского строения последнего; во-вторых, быстрая стадия реакции SN1 в значительной степени определяются зарядовым контролем, в котором роль основности существенно возрастают.
Таким образом, преимущественному замещению способствует менее разветвленная структура алкильной группы в исходном соединении, высокая нуклеофильность реагента по сравнению с его основностью, большая сольватирующая способность среды и умеренная температура. Очевидно, что отщеплению благоприятствуют противоположные условия.
Кроме рассмотренной конкуренции реакций реакции SN1 и Е1 могут сопровождаться побочными реакциями изомеризации. причина этих реакций состоит в том, что промежуточно образующийся карбкатион, прежде чем прореагировать с соответствующими нуклеофилом, способен к внутримолекулярной стабилизации за счет миграции к заряженному атому углерода отдельных атомов или групп.
В результате образуется новый карбкатион (или несколько карбкатионов), который при последующей реакции с нуклеофилом дает смесь продуктов замещения и отщепления. Подобные перегруппировки могут осуществляться следующими путями:
1. Миграция алкильных групп
Мигрирующая группа R перемещается вместе со связующей парой электронов и выполняет роль нуклеофила, внутримолекулярно атакующего реакционный центр. Поэтому его способность к миграции зависит от нуклеофильности и повышается при наличии в ней электронодонорных заместителей и с ростом их поляризуемости:
(CH3)3C-
> (CH3)2CH- > CH3CH2- > CH3-
2. Миграция атомов водорода (гидридный перенос)
Направление миграции атомов водорода определяется относительной стабильностью ионов карбония, часто находящихся в равновесии друг с другом. Поэтому преимущественно образуются третичные или вторичные продукты замещения или более разветвленные алкены. Особенно часто такие перегруппировки происходят при кислотнокаталитических превращениях спиртов. Так, при замещении гидроксила в бутаноле-1 на галоген получается на первичный, а вторичный бутилгалогенид:
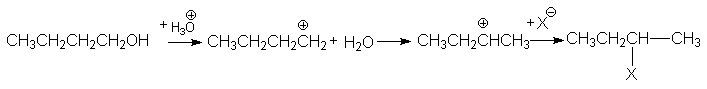
3. Перегруппировки, связанные с наличием в интермедиатах
нескольких реакционных центров
Причина образования таких центров обусловлена сопряжением вакантной орбитали карбкатиона с аллильной двойной связью или системой сопряженных двойных связей. Так, при гидролизе кротилхлорида образующийся карбкатион имеет два положительно заряженных реакционных центра. Поэтому при нуклеофильной атаке такого карбкатиона образуются два продукта замещения
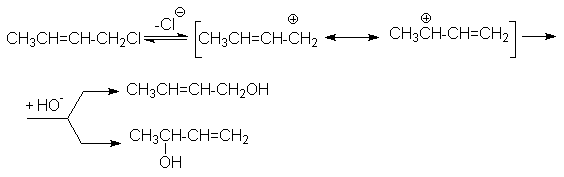
Для предотвращения всех типов изомеризации необходимо перевести реакцию из области SN1– Е1 в область SN2– Е2. Этому способствуют выбор апротонных неполярных растворителей, применение электрофильных катализаторов, снижение температуры и другие факторы, рассмотренные ранее.