Лекции
№ 41-42
АМИНЫ
План
Амины
можно рассматривать как производные аммиака, в котором атомы водорода замещены
на углеводородные радикалы.
1. Классификация, изомерия, номенклатура
В зависимости от числа углеводородных радикалов, связанных с атомом азота,
различают первичные, вторичные и третичные амины, а также четвертичные
аммониевые соли.
|
RNH2 |
RR/NH |
RR/R//N |
RR/R//
R///N+X- |
|
первичные
амины |
вторичные
амины |
третичные
амины |
четвертичные
аммониевые соли |
По типу гибридизации атома углерода, связанного с азотом выделяют следующие группы аминов.
- Соединения со
связью
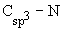 .
.
К этой группе относятся алкиламины, а также алкенил- и алкиниламины, в которых
кратная связь удалена от атома азота. Их объединяют под названием алифатические
амины. В состав этой группы входят также циклические амины, содержащие атом
азота в цикле, которые являются гетероциклическими соединениями.
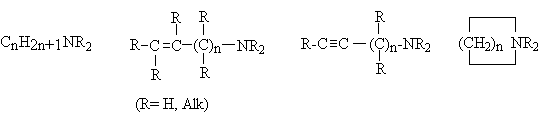
- Соединения со
связью
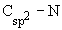 .
.
К
этой группе принадлежат производные алкенов с атомом азота у атома углерода,
образующего двойную связь – енамины (виниламины) и амины,
содержащие атом азота, связанный с ароматическим кольцом - ароматические
амины (ариламины).
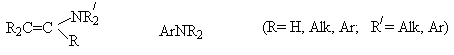
Названия аминов образуют, добавляя к слову амин названия связанных с атомом азота углеводородных радикалов.
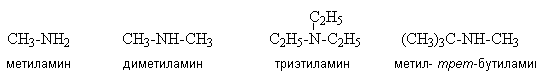
В другом варианте номенклатуры за основу названия принимают название родоначальной структуры (самой длинной углеродной цепи, непосредственно связанной с атомом азота) с добавлением суффикса “амин”.
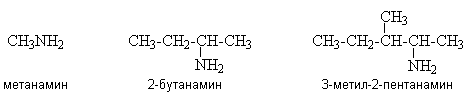
В этом случае вторичные и третичные амины называют как N-замещенные производные первичных аминов.
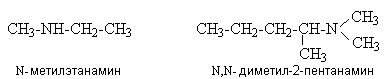
Если молекула содержит другие функциональные группы, обозначаемые в суффиксе, то аминогруппу обозначают префиксом “амино”.
![]()
Названия диаминов образуют от названий соответствующих двухвалентных радикалов или названия родоначальной структуры с добавлением суффикса “диамин”.
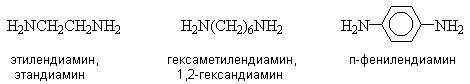
Многие ароматические амины имеют тривиальные названия.
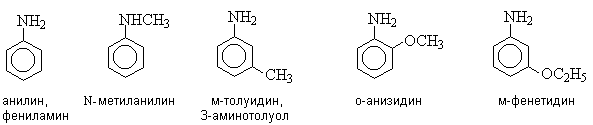
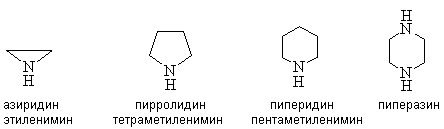
Циклические амины называют, используя номенклатуру гетероциклических соединений или, добавляя к названию двухвалентного углеводородного радикала суффикс “имин”.
Для аминов характерна изометрия углеродного скелета, изомерия положения
аминогруппы и изомерия между первичными, вторичными и третичными аминами.
2. Алифатические амины
2.1. Методы получения.
1) Алкилирование аммиака и аминов.
Аммиак взаимодействуют с алкилгалогенидами RX и другими алкилирующими реагентами (алкилсульфатами, диалкилсульфатами) с образованием на первой стадии соли алкиламмония, которая в равновесной реакции с избытком аммиака дает алкиламин. Алкиламин далее вступает в реакцию c алкилгалогенидом с образованием продукта диалкилирования и т.д. Таким образом последовательно образуются триалкиламин и соль тетраалкиламмония.
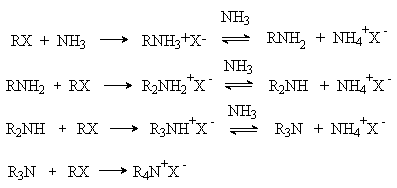
Реакция используется в основном для синтеза третичных аминов и тетраалкиламмониевых солей, так как первичные и вторичные амины, будучи более сильными нуклеофилами, чем аммиак, реагируют далее, сами предпочтительно атакуя субстрат. Приемлемые выходы первичных аминов получают при использовании большого избытка аммиака, а вторичных аминов – при большом избытке первичного амина.
Спирты алкилируют аммиак и амины в присутствии катализаторов дегидратации (Al2O3, SiO2) при 300-5000C. В этом случае также образуется смесь продуктов моно-, ди- и триалкилирования.
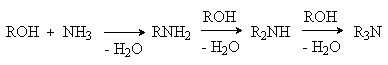
Метод используется для получения низших алифатических аминов в промышленности.
2) Синтез первичных аминов по Габриэлю
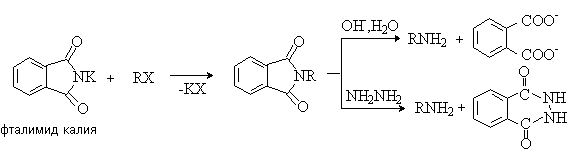
Алкилирование фталимида калия алкилгалогенидами с последующим щелочным гидролизом или гидразинолизом N-алкилфталимида позволяет получать первичные амины без примеси вторичных и третичных. Лучше использовать протекающий в мягких условиях гидразинолиз, приводящий к образованию не растворимого в реакционной среде циклического гидразида.
3) Восстановление азотсодержащих органических соединений.
Нитрилы при восстановлении дают первичные амины. В промышленности процесс осуществляют путем каталитического гидрирования.
![]()
В препаративных целях используют восстановление алюмогидридом лития.
![]()
Введение цианогруппы (например, путем нуклеофильного замещения) и ее восстановление – синтетический прием, позволяющий нарастить углеродную цепь на один атом С.
Амиды карбоновых кислот восстанавливаются до аминов алюмогидридом лития. Из соответствующих амидов могут быть получены первичные, вторичные и третичные амины.
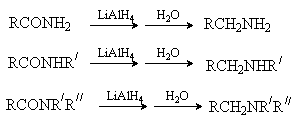
Восстановление азотсодержащих производных альдегидов и кетонов – оксимов и гидразонов – дает возможность превращения карбонильных соединений в первичные амины.
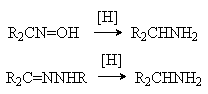
Для восстановления используют каталитическое гидрирование, комплексные гидриды металлов (LiAlH4).
Нитросоединения могут быть восстановлены до первичных аминов.
![]()
В качестве восстановителей чаще всего используют металл (Fe, Zn, Sn) и кислоту; алюмогидрид лития. В алифатическом ряду метод не находит широкого применения из-за ограниченной доступности алифатических нитросоединений по сравнению с ароматическими.
Восстановление азидов дает первичные амины.
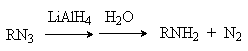
Исходные азиды легко могут быть получены из алкилгалогенидов или сульфонатов путем нуклеофильного замещения.
4) Восстановительное аминирование карбонильных соединений.
Взаимодействие альдегидов и кетонов с аммиаком в присутствии восстановителя приводит к первичным аминам.
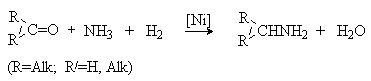
При использовании вместо аммиака первичного амина продуктом реакции будет вторичный амин.
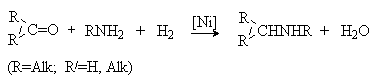
Процесс протекает через промежуточное образование имина с его последующим восстановлением в амин.
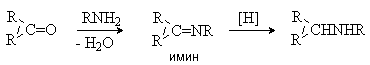
Восстановительное аминирование с использованием в качестве восстановителя муравьиной кислоты называют реакцией Лейкарта-Валлаха. В качестве реагентов можно использовать формиат аммония или соответствующие соли аминов.
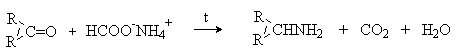
5) Синтез аминов путем перегруппировок.
Перегруппировка Гофмана:
RCONH2 + Br2
+ 2NaOH ® RNH2 + 2NaBr + CO2 + H2O
Перегруппировка Курциуса:
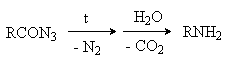
Реакции подробно рассмотрены ранее (см. лек. №36) В результате образуются
первичные амины без примеси вторичных и третичных. При этом происходит
укорочение углеродной цепи на один атом С.
2.2. Физические свойства и строение
Алифатические амины – бесцветные вещества с неприятным запахом. Низшие амины
– жидкости, хорошо растворимые в воде. По растворимости они превосходят спирты
с близкой молекулярной массой. Это объясняется образованием между амином и
водой водородных связей типа ![]() , прочность
которых сравнительно велика в силу высокой основности атома азота. Температуры
кипения и плавления у третичных аминов ниже, чем у первичных и вторичных с
примерно одинаковой молекулярной массой, что связано с ассоциацией последних за
счет образования межмолекулярных водородных связей.
, прочность
которых сравнительно велика в силу высокой основности атома азота. Температуры
кипения и плавления у третичных аминов ниже, чем у первичных и вторичных с
примерно одинаковой молекулярной массой, что связано с ассоциацией последних за
счет образования межмолекулярных водородных связей.
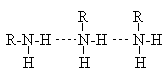
Однако эти межмолекулярные водородные связи слабее, чем у спиртов, по
причине меньшей полярности связи N-Н по сравнению со связью О-Н. Вследствие
этого амины имеют более низкие температуры кипения, чем спирты с близкой
молекулярной массой.
Амины имеют пирамидальное строение. Величины углов R-N-R близки к
тетраэдрическому – 106-1080. Считается, что атом азота находится в
состоянии sp3-гибридизации, а четвертым лигандом является
неподелённая пара электронов (“фантом”-лиганд).
Третичные амины с разными углеводородными радикалами должны быть хиральными,
так как их молекулы не имеют плоскости симметрии. Однако за счет быстрой
пирамидальной инверсии, которая представляет собой акт рацемизации, их
невозможно выделить или зафиксировать в оптически активной форме.
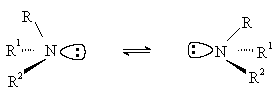
Четвертичные аммониевые соли в случае разных заместителей существуют в виде пары устойчивых энантиомеров.
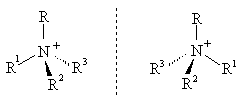
Спектральные характеристики.
В электронных спектрах аминов наблюдается поглощение в дальней УФ-области при 195-215 нм, что соответствует возбуждению неподеленной пары электронов азота (переход n® s* ).
В ИК-спектрах первичных и вторичных аминов наблюдаются полосы поглощения, связанные с валентными колебаниями связей N-H. Первичные амины характеризуются двумя полосами поглощения при ~3400 и ~3500 см-1, вторичные амины – одной полосой при ~3500 см-1.
В спектрах ПМР химический сдвиг протонов связи N-H находится в области 1-5 м.д. и значительно меняется в зависимости от концентрации, температуры и растворителя.
2.3. Химические свойства
Химическое поведение аминов определяется в основном наличием свободной пары электронов у атома азота, которая обусловливает их основные и нуклеофильные свойства. Реакции с участием связей N-H и N-C под действием оснований и нуклеофильных реагентов для аминов менее характерны.
Основные свойства
Алифатические амины являются одними из самых сильных незаряженных оснований
(![]() ~ 10 - 11). Их водные растворы имеют
щелочную реакцию.
~ 10 - 11). Их водные растворы имеют
щелочную реакцию.
R3N + H2O = R3NH+ + OH -
С неорганическими кислотами амины образуют соли, которые в большинстве случаев хорошо растворимы в воде.
R3N + HX = R3N+X -
Основность аминов зависит от их строения и природы растворителя. Сравнение
основности в водных растворах показывает, что алкиламины являются более
сильными основаниями, чем аммиак. Вторичные амины превосходят по основности
первичные. Такой ряд основности согласуется с электронодонорным влиянием
алкильных групп (+I-эффект), которое способствует делокализации положительного
заряда в сопряженной кислоте (ионе аммония) и тем самым стабилизирует её в
большей степени, чем свободный амин. Однако это не объясняет уменьшения
основности при переходе от вторичных аминов к третичным.
|
|
NH3 |
C2H5NH2 |
(C2H5)2NH |
(C2H5)3N |
|
|
9,25 |
10,80 |
11,09 |
10,85 |
Вероятно, такое снижение основности связано с сольватацией. Сольватация молекулами воды триалкиламмониевого катиона затруднена присутствием трех гидрофобных алкильных групп и снижением возможности образования водородных связей.

Это предположение подтверждается тем, что в газовой фазе и в малополярных
растворителях третичные амины превосходят по основности вторичные.
Нуклеофильные свойства
а) Алкилирование
Примеры реакций алкилирования обсуждались при рассмотрении методов получения аминов.
б) Ацилирование
2RNH2 + R/COX ® R/CONHR + RNH3X
2R2NH
+ R/COX ® R/CONR2 + R2NH2X
(X=Cl, OCOR/)
Ацилирование
аминов функциональными производными карбоновых кислот дает возможность получать
вторичные и третичные амиды из первичных и вторичных аминов соответственно.
Реакция подробно обсуждена ранее (лекция №36).
в) Взаимодействие с сульфонилхлоридами
Сульфонилхлориды взаимодействуют с аминами, давая сульфонамиды. Реакция с бензолсульфонилхлоридом лежит в основе пробы Гинсберга, позволяющей различать и разделять первичные, вторичные и третичные амины.
Сульфонамиды, образующиеся из первичных аминов, являются NH-кислотами и со щелочами дают растворимые в воде соли.
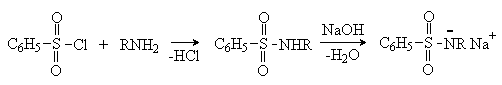
Вторичные амины дают сульфонамиды, которые не содержат кислого водорода и не растворяются в щелочах.
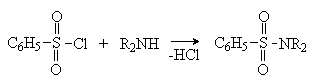
Третичные амины не реагируют.
г) Нитрозирование
Нитрозирование аминов происходит при взаимодействии с азотистой кислотой в кислой среде. Неустойчивую азотистую кислоту генерируют действием сильной кислоты на нитриты. Реакция протекает по-разному в зависимости от типа амина.
Первичные алифатические амины реагируют с образованием неустойчивых алкилдиазониевых солей, которые разлагаются с выделением газообразного азота и сложной смеси продуктов дезаминирования.
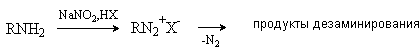
Образование солей диазония – сложный многостадийный процесс, который подробно будет рассмотрен на примере ароматических аминов.
Разложение катиона алкилдиазония приводит к образованию карбокатиона, который стабилизируется путем алкилирования присутствующих в реакционной среде нуклеофилов или путем отщепления протона с образованием алкена. Этим процессам может предшествовать изомеризация карбокатиона в энергетически более стабильный ион. Так, разложение катиона н-пропилдиазония в водном растворе наряду н-пропиловым спиртом дает изопропиловый спирт, а также продукт элиминирования - пропен.
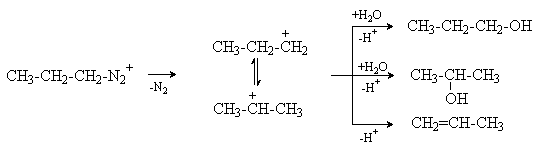
Со вторичными аминами азотистая кислота образует нерастворимые в реакционной среде нитрозамины.
R2NH + NaNO2
+ HCl ® R2N-N=O + NaCl + H2O
Третичные амины в сильнокислой среде при комнатной температуре с азотистой кислотой не реагируют.
Нитрозирование аминов препаративного значения не имеет. Аналитическое значение этих реакций заключается в возможности качественно различить первичные, вторичные и третичные амины.
д) Галогенирование
Первичные и вторичные амины реагируют с гипогалогенитами с образованием N-галогенаминов.
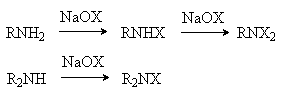
(X= Cl, Br)
N-галогенамины – сильные окислители и галогенирующие реагенты.
Окисление
Амины дают разнообразные продукты окисления, состав которых зависит от природы окислителя и строения амина.
Перекись водорода и надкислоты окисляют третичные амины до N-оксидов.
R3N + HOOH ® R3N+-O- + H2O
В случае первичных и вторичных аминов первоначально образующиеся N-оксиды перегруппировываются в производные гидроксиламина.
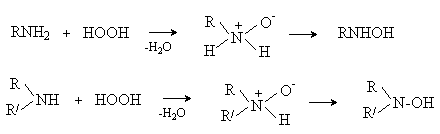
Такое окисление протекает сложно, так как гидроксиламины сами легко окисляются. В случае первичных аминов конечными продуктами окисления являются нитросоединения, например:
![]()
Первичные амины, в которых аминогруппа соединена с третичным атомом углерода, окисляются в нитросоединения перманганатом калия в водном ацетоне.
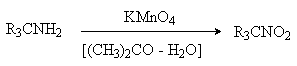
Амины, содержащие атомы водорода в a -положении, при действии сильных окислителей (KMnO4) дают смесь веществ, в которой преобладают карбонильные соединения. Процесс протекает через промежуточное образование иминов, которые при гидролизе дают альдегиды или кетоны.
![]()
(R=Alk; R/=H, Alk)
Кислотные свойства
Первичные и вторичные алифатические амины являются очень слабыми NH-кислотами (pKа~33-35). Их кислотные свойства проявляются при действии щелочных металлов или таких сильных основания, как металлоорганические соединения.
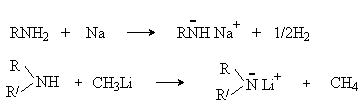
Образующиеся алкил- и диалкиламиды металлов – очень сильные основания. Диалкиламиды, содержащие вторичные или третичные алкильные радикалы (например, диизопропиламид лития), представляют интерес для органического синтеза как ненуклеофильные основания. Будучи сильными основаниями, они обладают низкой нуклеофильностью по причине стерических затруднений, возникающих при атаке электрофильных центров за исключением протона. Их используют в органическом синтезе для отрыва протона и генерирования карбанионов.
Расщепление гидроксидов тетраалкиламмония по Гофману
Гидроксиды тетраалкиламмония получают действием на галогениды оксида серебра.
2R4N+Br-
+ Ag2O + H2O ® 2R4N+OH-
+ 2AgBr
В растворах гидроксиды тетраалкиламмония полностью ионизированы и являются столь же сильными основаниями, как гидроксиды натрия и калия. При нагревании они претерпевают элиминирование с образованием алкена триалкиламина и воды.
RCH2CH2(CH3)3N+OH-® RCH=CH2 + (CH3)3N + H2O
При наличии в молекуле нескольких b -водородных атомов процесс протекает в направлении образования наименее замещенного алкена (по правилу Гофмана).
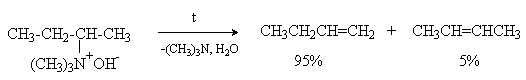
Причина такой ориентации при отщеплении состоит в карбанионном характере переходного состояния, что способствует отщеплению наиболее кислого протона.
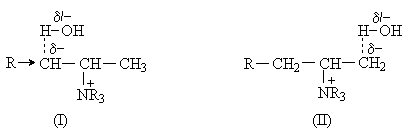
При протекании процесса по механизму Е2 с “Е1СВ-подобным” переходным состоянием на атоме углерода возникает частичный отрицательный заряд. Переходное состояние (I), предшествующее образованию продукта по правилу Зайцева, оказывается дестабилизированным за счет +I-эффекта алкильных групп. В результате процесс преимущественно направляется через наименее дестабилизированное переходное состояние (II), ведущее к продукту элиминирования по Гофману.
3. Енамины
Енамины (виниламины) устойчивы в том случае, если при атоме азота нет атомов водорода. Такие енамины можно рассматривать как азотистые аналоги виниловых эфиров. В противном случае енамины нестабильны и перегруппировываются в имины, подобно тому, как енолы изомеризуются в карбонильные соединения.
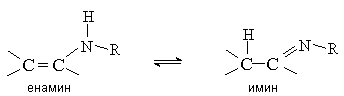
3.1. Методы получения
Основной метод получения енаминов – взаимодействие карбонильных соединений со вторичными аминами в присутствии кислотных катализаторов и средств, связывающих воду.

Реакция протекает по общему для присоединения азотистых оснований к карбонильной группе механизму.
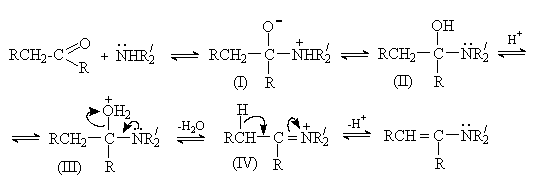
Отщепление воды от интермедиата (III) приводит к образованию иммониевого иона (IV), который при отсутствии водорода у атома азота стабилизируется путем отщепления протона от b -углеродного атома.
3.2. Строение
Молекула енамина представляет собой р-p -сопряженную систему, строение которой можно отразить набором двух резонансных структур.
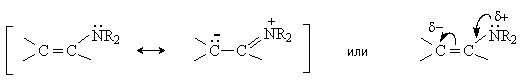
Таким образом, молекула енамина содержит два нуклеофильных центра – атом азота и атом углерода в b -положении, который несет частичный отрицательный заряд.
3.3. Химические свойства
а) Протонирование и гидролиз
Енамины являются слабыми основаниями. Их протонирование может протекать как по атому азота, так и по атому углерода. Образующаяся при протонировании по b -углеродному атому соль иммония гидролизуется, давая исходное карбонильное соединение и вторичный амин.
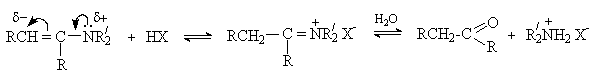
Гидролиз енаминов – процесс, обратный их образованию, и протекает по такому же механизму.
б) Алкилирование
Алкилирование енаминов алкилгалогенидами и другими алкилирующими реагентами протекает, как правило, по b -углеродному атому. Последующий гидролиз иммониевой соли приводит к карбонильному соединению.
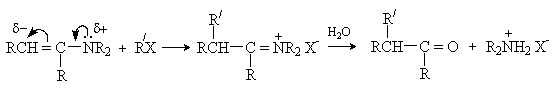
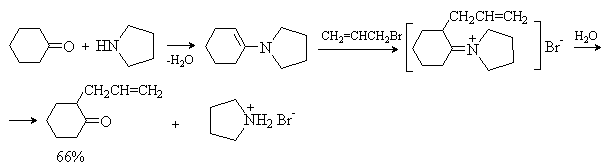
Последовательность превращений – получение енамина из карбонильного соединения, алкилирование, гидролиз приводит к алкилированию исходного карбонильного соединения по a -положению и носит название реакция Сторка. Этот метод имеет преимущества перед алкилированием кетонов, так как требует более мягких условий и дает в основном продукты моноалкилирования.
Для проведения этой реакции чаще всего используют циклические амины – пирролидин, пиперидин, морфолин. Лучшие результаты достигаются при использовании активных галогенидов – аллил- и бензилгалогенидов, a -галогензамещенных производных простых и сложных эфиров. Например:
в) Ацилирование
При действии галогенангидридов и ангидридов кислот енамины дают продукты С-ацилирования. Последующий гидролиз приводит к дикарбонильному соединению.
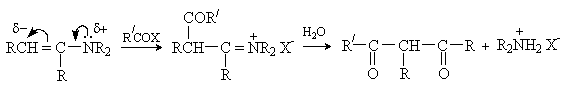
Таким образом, последовательность реакция – получение енамина из карбонильного соединения, ацилирование, гидролиз – метод получения b -дикарбонильных соединений. Например:
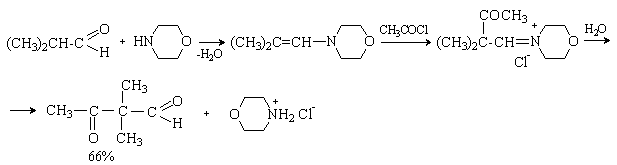
4. Ароматические амины
4.1. Методы получения
1) Восстановление ароматических нитросоединений
![]()
Для восстановления в препаративных целях используют металл (Fe, Zn, Sn) и кислоту, соли металлов в низших степенях окисления (SnCl2, TiCl3), сульфиды щелочных металлов, в промышленности применяют в основном каталитическое гидрирование. См. также лекцию №40.
2) Алкилирование
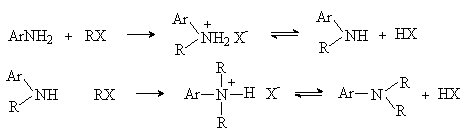
Реакция аналогична алкилированию алифатических аминов. В качестве алкилирующих реагентов используют алкилгалогениды, алкилсульфаты, спирты.
3) Арилирование
Галогенарены реагируют с аммиаком и аминами в жестких условиях. Процесс катализируется медью и ее соединениями.
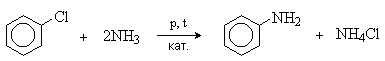
Реакция замещения галогена протекает легче при наличии в орто- и пара-положениях электроноакцепторных групп (NO2, CN).
Галогенарены взаимодействуют с ариламинами в присутствии меди с образованием диариламинов (реакция Ульмана).
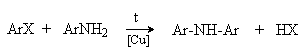
4.2. Физические свойства и строение
Ароматические амины – бесцветные жидкости или твердые вещества. При хранении быстро темнеют вследствие окисления кислородом воздуха.
Аминогруппа и ароматическое кольцо образуют сопряженную систему. Аминогруппа проявляет электронодонорные свойства за счет +М-эффекта.
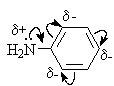
Ароматические амины обладают сильными электронодонорными свойствами, на что указывают низкие энергии ионизации (для анилина 7,7 эВ, для фенола 8,4 эВ).
4.3. Химические свойства
Для ариламинов характерны реакции с электрофильными реагентами. Местом атаки электрофила может быть атом азота или ароматическое кольцо.
Основные свойства
Ароматические амины обладают меньшей основностью, чем алифатические амины и
аммиак (![]() ~ 3 – 5). Причина низкой основности
ариламинов – стабилизация свободного амина за счет делокализации неподеленной
пары электронов азота по ароматическому кольцу и потеря энергии стабилизации
при нарушении сопряженной системы в результате протонирования.
~ 3 – 5). Причина низкой основности
ариламинов – стабилизация свободного амина за счет делокализации неподеленной
пары электронов азота по ароматическому кольцу и потеря энергии стабилизации
при нарушении сопряженной системы в результате протонирования.
Дифениламин и трифениламин имеют еще большие возможности для делокализации пары электронов азота, что приводит значительному снижению основности. Трифениламин практически не обладает основными свойствами.
Введение заместителей в ароматическое кольцо влияет на основность амина.
Влияние заместителей проявляется в соответствии с их электронными эффектами.
|
|
|
|
|
|
п-CH3C6H4NH2 |
5,1 |
п-O2NC6H4NH2 |
1,0 |
|
C6H5NH2 |
4,6 |
(C6H5)2NH |
0,78 |
|
м-O2NC6H4NH2 |
2,5 |
|
|
Реакции с С-электрофилами
Важнейшими реакциями этого типа являются алкилирование и ацилирование, которые протекают по атому азота и аналогичны реакциям алифатических аминов. Ароматические амины менее реакционноспособны из-за меньшей основности атома азота.
Реакции ароматического электрофильного замещения
а) Галогенирование
Аминогруппа является сильным активирующим заместителем и ориентантом I рода. Галогенирование свободных аминов протекает очень легко и часто приводит к полигалоидпроизводным. Например, анилин при действии бромной воды мгновенно превращаются в нерастворимое 2,4,6-трибромпроизводное.
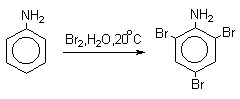
Для получения моногалогенпроизводных активирующее действие аминогруппы снижают путем ацилирования. После снятия ацильной защиты путем гидролиза получают свободный амин.
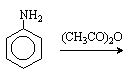
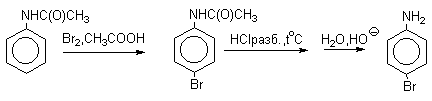
б) Нитрование
При нитровании нитрующей смесью амины окисляются. Кроме того, из-за солеобразования по аминогруппе образуется м-изомер (-NH3+ - ориентант II рода).
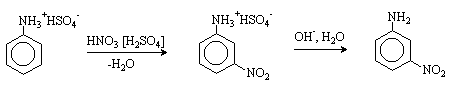
Для введения нитрогруппы в орто- или пара-положение к аминогруппе последнюю защищают ацилированием. Варьируя условия реакций (температуру, нитрующие агенты), можно проводить нитрование региоселективно.
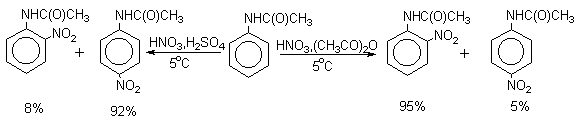
После снятия ацетильной защиты получают свободные орто- и пара-нитроанилины.
в) Сульфирование
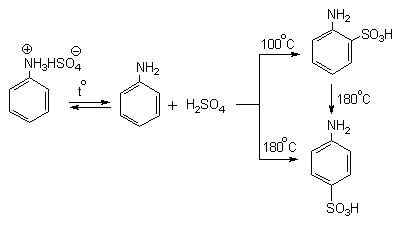
Сульфированием ароматических аминов получают аминосульфокислоты. В 90-100%-ной серной кислоте или олеуме амин полностью находится в протонированной форме. Аммониевая группа NH3+как сильный электроакцепторный заместитель вызывает резкое замедление реакции сульфирования и ориентирует замещение в мета-положение.
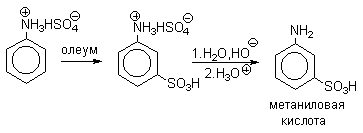
Для получения орто- и пара-аминобензолсульфокислот используют “метод запекания”. Процесс осуществляют при длительном нагревании гидросульфатов ароматических аминов при 100-200оС в сухом виде или в высококипящих растворителях. При температуре около 100оС образуется практически чистый орто-изомер (ортаниловая кислота, продукт кинетического контроля), а при 180-200оС - пара-изомер (сульфаниловая кислота, продукт термодинамического контроля).
Нитрозирование
Первичные ароматические амины с азотистой кислотой образуют относительно устойчивые соли арилдиазония.
ArNH2 + NaNO2
+ 2HCl ® ArN2+Cl- +
NaCl + 2H2O
Эту реакцию называют диазотированием (см.лек. №43).
Вторичные ароматические амины при нитрозировании дают N-нитрозамины.
ArNHR + NaNO2 +
HCl ® Ar-N(R)-N=O + NaCl + H2O
Третичные ариламины дают продукты нитрозирования в пара-положение ароматического кольца.
![]()