Лекция 3
Второе начало термодинамики.
Энтропия и методы её
определения.
План
1. Внутренняя
неупорядоченность макросистемы и термодинамическая температура.
2. Второе
начало термодинамики.
3. Третье
начало термодинамики.
4. Термодинамические потенциалы, как критерии
самопроизвольного протекания процесса.
По
величине внутренней энергии и энтальпии нельзя судить находится ли данная система в равновесном
состоянии или нет, а также
ничего нельзя сказать о возможности протекания процессов в этих системах.
Поэтому для этой цели нужна другая функция состояния, которая может служить
критерием самопроизвольного протекания необратимых процессов.
Наиболее характерной чертой
всякого макроскопического тела является то, что пространственное расположение и
движение образующих его микрочастиц в большей или меньшей степени являются
беспорядочными. Это беспорядочное расположение частиц в макротеле создается
само собой, тогда как упорядоченное в хаотическом расположении частиц требует затраты работы. В
идеале, за количественную меру беспорядка в системе сложно взять работу по
«наведению» идеального порядка. Однако можно дать и более общее и более точное
определение меры беспорядка в системе, не требующее её перевода в идеально
упорядоченное состояние.
Количественная мера
внутренней неупорядоченности системы в термодинамике называется энтропией-S.
Произвольное макротело изолированное от среды в силу атомно-молекулярной
природы по истечению какого-то времени само собой придет в состояние
термодинамического равновесия и будет прибывать в нем сколь угодно долго. С
точки зрения внутренней неупорядоченности, всякий процесс релаксации – это
переход от состояния с большей упорядоченностью в состояние с меньшей
упорядоченностью. В изолированной системе этот процесс ограничен пределами,
когда достигается максимальная неупорядоченность, поэтому процесс релаксации в изолированной системе всегда ведет к росту
энтропии. Это общее свойство энтропии любых макроскопических тел, находящихся в
изоляции, называется принципом максимума энтропии.
В термодинамике, с её
феноменологическим подходом, величина энтропии введена формально. Связь
энтропии с мерой внутренней неупорядоченности макротел была установлена позднее
в статистической механике.
Увеличение энтропии
- это результат неравновесности протекания
процесса, который сопровождается рассеиванием энергии. Чем больше рассеивается
энергии, тем больше применение энтропии.
Если в системе протекает
обратимый процесс, то внутренняя неупорядоченность закрытых систем может
меняться только тогда, когда система обменивается энергией со средой в форме
теплоты, а не в форме работы. Если система поглощает энергию из вне и получает
ее в форме теплоты, то возрастают одновременно 2 термодинамические функции,
внутренняя энергия и энтропия. Таким
образом, внутренняя энергия является монотонно возрастающей функцией энтропии.
Следовательно, для простых равновесных систем:
U =
U (V,S)
(3-1)
Для (3-1) мы можем найти полный дифференциал внутренней энергии.
dU = (![]() )S * dV + (
)S * dV + (![]() )V * dS (3-2)
)V * dS (3-2)
Если сопоставить уравнение (3-2) с выражением 1–го начала термодинамики:
dU = dQ – pdV (3-3)
то можно заметить:
(![]() )V * dS = dQ, а (
)V * dS = dQ, а (![]() )S =
- p
)S =
- p
Обратим
внимание на то, что (![]() )V обладает
всеми признаками температуры, поэтому (
)V обладает
всеми признаками температуры, поэтому (![]() )V
= Т, а (3-3) можно записать
)V
= Т, а (3-3) можно записать
dU = TdS – pdV
(3-3` )
Второе начало термодинамики.
Ф. Кларуфиус, на основе анализа работы тепловой машины, работающей по циклу Карно, показал, что для обратимых процессов в макросистемах приведенная теплота обладает свойствами функции состояния:
dS = ![]() (3-4)
(3-4)
Эта
функция состояния интересна тем, что равенство (3-4) выполняется только для
обратимых процессов, а в случае необратимых процессов имеет место неравенство:
dS > ![]() (3-4`)
(3-4`)
Соотношения
(3-4) и (3-4`) представляют общую формулировку второго начала термодинамики в
аналитической виде: дифференциал энтропии не убывает.
Рассмотрим
важнейшие следствия из этой общей формулировки:
1.
Второе начало термодинамики позволяет определить изменение энтропии на
основании калориметрических данных. Таким образом, энтропия, как и энтальпия и
внутренняя энергия является термической константой.
Действительно,
∆ S(p) = 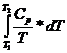 =
= 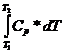 lnT
(3-5)
lnT
(3-5)
а для фазовых переходов (плавление,
испарение):
∆ S(p) = ![]() (3-6)
(3-6)
2. При
адиабатической изоляции применение энтропии не отрицательно.
Q = 0 → ∆ S ![]() 0 (3-7)
0 (3-7)
Это служит критерием
самопроизвольного процесса в адиабатической системе.
3. Соотношение (3-7)
справедливо для любых изолированных систем. Поэтому можно сформулировать энтропийный
принцип: энтропия изолированной системы при протекании в ней обратимых
процессов не изменяется, а при протекании необратимых процессов – возрастает.
Третье начало термодинамики.
II
второе начало термодинамики позволяет определить только применение энтропии
тел. Однако, обширный материал экспериментальных работ В. Нернста и других
исследователей позволил сделать вывод, что при понижении температуры энтропия
уменьшается. Причем, чем ближе температура а 0 К, тем ниже энтропия. Этот факт
лежит в основе третьего начала термодинамики. При равновесном приближении к
абсолютному нулю температуры энтропия макроскопических тел стремится к нулю.
![]() S = 0 (3-8)
S = 0 (3-8)
Соответственно
при неравновесном понижении температуры
система остается в «замороженном» метастабильном состоянии. Причем, это
фактически неизбежно в реальном опыте. По мере понижения температуры,
возрастает время релаксации до бесконечности. Поэтому из III
начала термодинамики вытекает в качестве следствия принцип недостижимости абсолютного нуля температур.
Другим
важным следствием третьего начала термодинамики является заключение о характере
температурной зависимости теплоемкости при температурах, близких к 0 К. Это
позволяет определять абсолютные энтропии.
Термодинамические потенциалы
Рассмотрим
следствия, вытекающие из совместного применения I и II начала термодинамики к
произвольной системе, взаимодействующей с окружающей средой:
TdS ![]()
![]() = dU +
= dU + ![]() (3-9)
(3-9)
I начало термодинамики
применимо как к обратимым, так и к необратимым процессам. Однако, при замене ∂Q на TdS для необратимых процессов выполняется неравенство.
Предположим
в системе совершается работа связанная с действием различных сил не связанных с
механической работой, то выражение для этого случая в необратимых процессах
выражается:
- ![]() > dU + pdV - TdS (3-10)
> dU + pdV - TdS (3-10)
Из (3-10) следует:
1) при V = const, S = const - ![]() > dU
> dU
2) при p = const, S = const - ![]() > dU + pdV = d (U +pV) = dH
> dU + pdV = d (U +pV) = dH
3) при p = const, T= const - ![]() > dU + dpV – dTS = d (U + pV - TS)= = d (H - TS)
> dU + dpV – dTS = d (U + pV - TS)= = d (H - TS)
4) при V = const, T= const - ![]() > dU – TdS = dU – dTs = d (U - TS)
> dU – TdS = dU – dTs = d (U - TS)
Исходя из выражения (3-10) с учетом того, что
самопроизвольный процесс – это процесс который сам способен производить
положительную работу, то для случаев 1) - 4) можно записать условия самопроизвольного
протекания необратимого процесса:
d П(x,y) < 0
(3-11)
где П(х,у) это U(V,S)
или Н(Р,S) или выражения под знаком дифференциала для случаев
3), 4). Эти функции получили специальное название, потому что они имеют
практический интерес (свободная энергия Гиббса (G) и Гельмготца
F))
G = H – TS
(3-12)
F = U – TS
(3-13)
Очевидно, что ∆G(P,T) и ∆F(V,T) могут быть определены из
эксперимента, поскольку обеспечить р=соnst и Т= соnst или V= соnst и Т= соnst в эксперименте не представляет труда.
Выражение (3-11) определяет фактически
направленность необратимого процесса: при самопроизвольном процессе убывает термодинамический
потенциал.
Итак, нами установлено, что
П(х,у) = 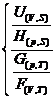
Все
эти функции при постоянстве своих естественных переменных убывают при
самопроизвольном необратимом процессе и служат критерием направленности этих
процессов.
У этих функций своим естественным
переменным есть еще одно замечательное свойство. Взяв производные от этих
функций по естественным переменным можно выразить все термодинамические
параметры, т.е. охарактеризовать термодинамическую систему в целом. Поэтому все
термодинамические потенциалы являются характеристическими функциями.
Поскольку с точки зрения математики
безразлично, что взять в качестве аргумента, то очевидно характеристической
функцией может быть и V = f (U,S) = ψ (F,Т) или температура Т = ψ (G,Р) = ψ (F,V).
Поэтому, строго говоря, характеристическая функция – это более общее понятие,
чем термодинамический потенциал.
f (p, Т, V)
= 0
(1)
рV
= n R T
(1.2)
δQ = dU + δA (1.3)
Q = ∆U
+ A
(1.3′)
Q = ∆U + p ∆V + A′ = ∆U + ∆ pV + A′
= ∆ (U + pV) + A′ (1.4)
Н = U
+ pV
(1.4′)
Из выражения (1.4) следует, что
при A′ = 0:
QV = ∆U, а Qp = ∆H (1.5)