Лекция 30. Мономолекулярные реакции
Оглавление
1.3.
Теория РРКМ (Рейса-Ратснерма–Касселя-Маркуса)
В истории
химической кинетики мономолекулярные реакции занимают особое место. Ещё Вант -
Гофф изучая разложение AsH3 и PH3 в газовой фазе отнёс эти реакции к мономолекулярным.
Однако, позднее было обнаружено, что разложение этих веществ протекает на
стенке, а не в объеме (реакция Марша). В начале 20-х годов XX в. Первый порядок был открыт для разложения N2O5, но механизм оказался сложнее. Только в 1922 была
обнаружена первая мономолекулярная газовая реакция – изомеризация циклопропана
в пропилен.
Широко
известная теория ТАС, однако не давала представлений о способе активации частиц
в мономолекулярных реакциях.
В 1922 г.
Предложил оригинальную гипотезу о путях активации частиц в мономолекулярных
реакциях.
В
соответствии со схемой Линдемана первая стадия взаимодействия – бимолекулярное
столкновение частицы А с любой частицей в реакционной смеси:
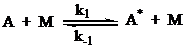
Затем
часть активированных частиц А* переходят в продукт реакции:
![]()
Поскольку
концентрация А* мала её можно считать стационарной и используя метод
стационарных состояний:
![]()
При записи
этого выражения предполагалось, что СА = СМ. Таким образом,
СА легко находятся:
![]()
Таким
образом, скорость накопления продукта:
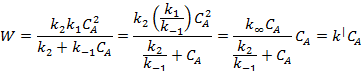
где
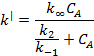
Общий вид k′=f(CA)
приведем на рисунке:
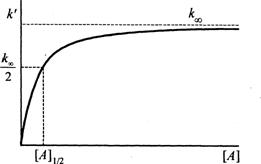
Зависимость
наблюдаемой константы скорости мономолекулярной реакции от концентрации
реагирующего вещества.
Такие
экспериментальные зависимости получили неоднократное подтверждение. Однако
количественный анализ констант скорости в модели Линдемана неоднократно
обнаруживал существенные расхождения теоретических и экспериментальных величин.
Хиншельвуд
одним из первых понял, что рассматривать бесструктурные модели реагирующих
частиц некорректно.
Главная
идея Хиншельвуда заключалась в том, что наряду с некоторой энергией, которая в
результате неупругих столкновений переходит во внутреннюю колебательно-вращательную
эти молекулы сами «активированы» за счёт собственного запаса внутренней
энергии. Это предположение внесло существенные коррективы в схему Линдемана.
Если в схеме Линдемана активация протекает путём повышения кинетической энергии
по центров над пороговым уровнем энергии εа, то в модели
Хиншельвуда порог СА* может быть повышен и путём перераспределения
собственной энергии колебания.
Для
процессов дезактивации и химического превращения константы этих процессов в
теории Хиншельвуда принимаются постоянными.
Итак, по
Хиншельвуду после бимолекулярного столкновения частица А активируется в узко
интервале энергий [ε, ε+dε]:
![]()
Если при
столкновении участвуют f –
колебательных степеней свободы, то полной колебательной и поступательной
энергии в процессе отвечают:
![]()
квадратичных члена.
Вероятность
попадания энергии молекулы, которая выражается f- квадратичными членами, в интервале ε, ε+dε
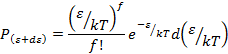
Вероятность
того, что ![]()
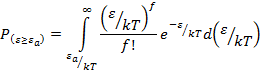
Если ![]() , то интеграл
приближённо равен:
, то интеграл
приближённо равен:
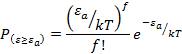
Таким
образом, учёт внутренних степеней свободы приводит к увеличению квадратичных
членов и доля молекул с ![]() редко вырастает
и при
редко вырастает
и при ![]() может быть
найдена:
может быть
найдена:
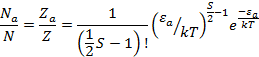
Очевидно
при S=2 это выражение переходит в больцмановский множитель.
Схема
Линдемана модифицированная Хиншельвудом приобретает следующий вид:
P – высокое ![]()
P – низкое ![]()
Зависимость
эффективной константы скорости k1, от давления получается комбинированием уравнения
Линдемана и выражения Хиншельвуда:
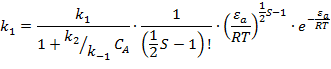
1.3. Теория РРКМ (Рейса-Ратснерма–Касселя-Маркуса)
В 1927 г.
Райнс и Ратснергер и в 1928 г. Кассель внесли новый элемент в теорию
мономолекулярных реакций. Они записали схему Линдемана в виде:
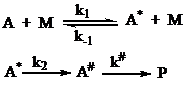
После
соударения А# имеет ![]() и в принципе
может перейти а A#, но этот процесс чисто вероятностный. Предполагается,
что энергии, аккумулируемые на каких-то
осцилляторах молекулы >> kT. Переход k2
предполагает сосредоточение энергии на одном осцилляторе (т.е.сосредотачивается
на колебательный моде соответсвует одной из связей, которая рвется). Исходя из
этой теории:
и в принципе
может перейти а A#, но этот процесс чисто вероятностный. Предполагается,
что энергии, аккумулируемые на каких-то
осцилляторах молекулы >> kT. Переход k2
предполагает сосредоточение энергии на одном осцилляторе (т.е.сосредотачивается
на колебательный моде соответсвует одной из связей, которая рвется). Исходя из
этой теории:
![]()
где ![]() - критическая энергия А*
- критическая энергия А*
а ![]() - энергия А*
- энергия А* ![]() за счет которой идет переход
за счет которой идет переход ![]()
f – общее число
осцилляторов в активированной частице.
Эта теория
предсказывала значния ![]() для всех
мономолекулярных реакций
для всех
мономолекулярных реакций ![]() тогда как при
распаде многих органических молекул на радикалы
тогда как при
распаде многих органических молекул на радикалы ![]() . Это требовало объяснений.
. Это требовало объяснений.
В 1951 –
1952 гг. Маркус видоизменил и дополнил модель РРК с использованием квантово –
механического подхода.
По Маркусу
![]() молекулы А*
больше
молекулы А*
больше ![]() , т.е. разности нулевых энергий колебательных уровней
активированного комплекса и исходной
молекулы А. Однако часть от
, т.е. разности нулевых энергий колебательных уровней
активированного комплекса и исходной
молекулы А. Однако часть от ![]() не
перераспределятся между неактивными степенями свободы
не
перераспределятся между неактивными степенями свободы ![]() . У активированного комплекса
. У активированного комплекса ![]() исключена из
распределения и
исключена из
распределения и ![]() .
.
Применяя
методы статистической термодинамики Маркусом было получено уравнение, с
точностью до множителя совпало с уравнением, полученным в рамках теории
переходного состояния. Это практически укрепило эти две теории. Кроме того,
теория РРКМ дала объяснение большим значениям Р для многих мономолекулярных
реакий.
Однако при
расчёте констант мономолекулярных реакций необходима модель А* и ![]() для выбора
частот и колебательных под активных и неактивных. Выбор этот зачастую
неоднозначен и опирается в основном на постулат Хэммонда (1955) который
утверждает, что для сильно экзотермических реакций структура
для выбора
частот и колебательных под активных и неактивных. Выбор этот зачастую
неоднозначен и опирается в основном на постулат Хэммонда (1955) который
утверждает, что для сильно экзотермических реакций структура ![]() близка к А
наоборт для сильно эндотермических она близка к Р.
близка к А
наоборт для сильно эндотермических она близка к Р.