Гидроксилпроизводные углеводородов. Спирты и фенолы, их свойства, способы
получения.
Гидроксилпроизводные следует рассматривать как результат замещения атомов
водорода в углеводороде на гидроксильную группу.
Гидроксилпроизводные могут быть подразделены по типу
атомов углерода, связанных с гидроксильной группой:
1) гидроксилпроизводные С (SP3) - ОН;
2) гидроксилпроизводные С (SP2) - ОН.
Гидроксилпроизводные С (SP) - ОН неизвестны, известны только их
производные - эфиры С(SP) - OR.
Гидроксилпроизводные со связью С (SP3) -
ОН.
К ним относятся:
а) гидроксилпроизводные
алканов и циклоалканов - алканолов и циклоалканолов;
б) гидроксилпроизводные
алкенов, алкинов, циклоалкенов,
если С=С и СºС - группы отделены от группы ОН по крайней мере одним
атомом углерода в состоянии sp3, например:
СН2 = СН ¾ СНОН СН º С ¾ СН2 ¾ СН2ОН
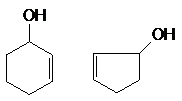
в) гидроксилпроизводные
алкиларенов в которых
гидроксильная группа находится в алкильном заместителе
ArCH2OH Ar(CH2)nOH
Алканолы – результат замещения атомов водорода в алканах на ОН
- группы.
Различают одноатомные и многоатомные алканолы. Последние представляют собой результат замещения
атомов водорода в алканах при различных углеродных атомах,
например:
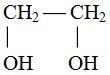 НОСН2СНОНСН2ОН
НОСН2СНОНСН2ОН
Эмпирическая формула одноатомных спиртов
СnH2n+2O.
Изомерия и номенклатура одноатомных алканолов.
В зависимости от типа углеродного атома,
с которым связана гидроксильная группа, различают первичные, вторичные и третичные
алканолы (спирты):
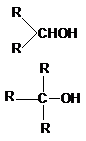
RCH2OH – первичные.
Согласно заместительной номенклатуре, углеродные атомы
цепи нумеруют таким образом, чтобы гидроксильная группа обозначалась меньшим
номером. Если гидроксильная группа старшая, то ее обозначают суффиксом “-ол”. Если в молекуле имеются
более старшие группы ( > С = О, ¾ СООН), то гидроксильную группу обозначают префиксом “гидрокси-“
(иногда “окси-”). По радикально-функциональной номенклатуре алканолы
называются спиртами, например:
Изомерия алканолов
определяется местонахождением гидроксильных групп и разветвлением углеродной
цепи. Начиная с бутанола -2, имеет место также стереоизомерия,
т.к. появляется хиральный атом углерода.
1. Гидратация алкенов
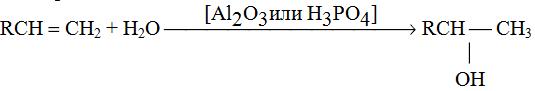
2. Гидроборирование алкенов
RCH = CH2 ![]() ® RCH2CH2OH
® RCH2CH2OH
3. Гидролиз галогеналканов
2RCl
+ Na2CO3 +H2O ® 2ROH + 2CO2
+ 2NaCl
4. Гидрирование карбонильных соединений.
а) Восстановление водородом
RCHO + H2 RCH2OH
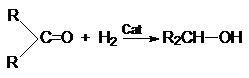
RCOOR¢ + H2 RCH2OH
+ R¢OH
Катализаторами этих процессов являются Ni, Pt, Pd.
б) Восстановление алкоголятами
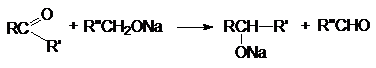
Восстановление осуществляется благодаря
переносу гидрид - аниона от алкоголята к
в) Восстановление неорганическими
гидридами.
Исключительно важное препаративное
значение при восстановлении карбонильных соединений приобрели неорганические
гидриды – литий алюминий-гидрид LiAlH4 и натрий
боргидрид NaBH4. Эти
реагенты находят широкое применение, особенно в случае неустойчивых и
дорогостоящих карбонильных производных. Хорошим примером может служить восстановление
циклобутанона в циклобутанол.

или суммарно:
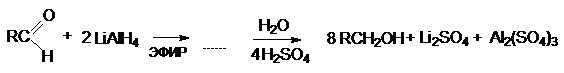
5. Взаимодействие карбонильных
соединений с реактивом Гриньяра
а) CH2 = O + RMgX ® R¾CH2 ¾OMgX RCH2OH
+ Mg(OH)X
Можно видеть, что используя в качестве реагента формальдегид получают первичные спирты, содержащие на один
атом углерода больше, чем исходное карбонильное соединение.
б) В то же время, конденсация альдегидов (кроме
формальдегида) с реактивами Гриньяра с последующим
гидролизом приводит к образованию вторичных спиртов.
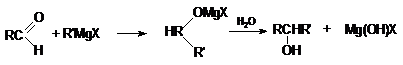
в) В свою очередь, конденсация кетонов с
реактивами Гриньяра и последующий гидролиз приводит к
образованию третичных спиртов.
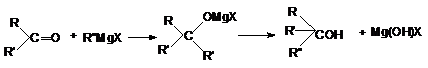
6. Синтез спиртов из синтез-газа
СO + 2H2 CH3OH
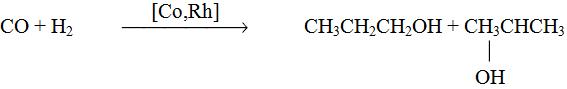
Физические свойства алканолов.
Алканолы являются бесцветными жидкостями или кристаллическими
веществами с характерным запахом. Первые члены ряда имеют приятный запах, для бутанолов
и пентанолов запах становится неприятным и раздражающим.
Высшие алканолы имеют
приятный ароматический запах.
Отличительная черта алканолов
- более высокая температура кипения, чем для соответствующих хлор-, бром- и иодалканов, несмотря на то,
что молекулярные массы галогеналканов выше.
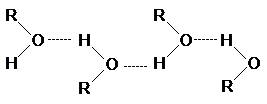
Это явление обусловлено сильными
межмолекулярными взаимодействиями в спиртах за счет образования водородных
связей.
Алканолы содержат две полярные связи Сd+¾ Оd- и Od+¾ Hd-. Диполи этих связей направлены навстречу друг другу.
Однако результирующий дипольный момент направлен от группы R.
Потенциал ионизации алканолов
ниже, чем у воды, что обусловлено +I - эффектом алкильной группы.
Спектры поглощения.
Алканолы в электронных спектрах поглощения являются “прозрачными”.
Слабое поглощение наблюдается только в дальней УФ - области (170-180нм), что
связано с переходом n ® s * неподеленной пары
электронов кислорода.
В ИК - спектрах
характерные колебания связи ОН в разбавленных растворах углеводородов или галогенуглеводородов наблюдаются в области 3580 - 3650 см-1. В
концентрированных растворах образуются водородные связи
О ¾ H ×××× О,
поэтому валентные колебания связи О ¾ Н смещается в область 3200-3500 см-1.
В спектрах ПМР сигнал группы ОН
наблюдается в широком интервале d=2 - 4,5 м.д. в зависимости от типа растворителя и
концентрации вещества. Характерные сигналы протонов Н ¾
С ¾
О при d
= 3,5 - 3,8 м.д.
Спирты являются слабыми ОН-кислотами, которые проявляются во взаимодействии с
сильными основаниями.
ROH + ОН- = RO- + H2O
Это равновесие существенно смещено в
левую сторону, т.к. RO- дестабилизирован из-за положительного индуктивного эффекта алкильной
группы. Обычно для получения алкоголятов применяют реакцию активного металла с
безводным спиртом.
2ROH
+ 2Na ® 2RONa + H2
6ROH
+ 2Al ® 2Al(OR)3 +
3H2
Высокая основность алкоголятов используется для успешного осуществления
реакций дегидрогалогенирования и генерирования карбанионов и других анионов, играющих ключевую роль в
реакциях конденсации органических соединений
![]()
В тоже время алкоголят-анионы
являются достаточно сильными нуклеофилами, что
позволяет использовать их в качестве реагентов в некоторых важных реакциях
нуклеофильного замещения, например, в синтезе простых и сложных эфиров.
RHal + R¢ONa ® R¢OR + NaHal
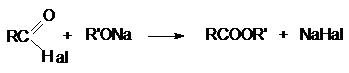
Неподеленная электронная пара на кислородном атоме гидроксильной
группы обуславливает возможность взаимодействия его с электрофильными
реагентами с образованием донорно-акцепторной связи, при этом кислородный атом приобретает положительный заряд и образуются оксониевые соединения:
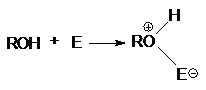
где E - кислота Льюиса, например BF3,
AlCl3, PCl3, SOCl2, TiCl4,
SnCl4, H+, NO2 и др.
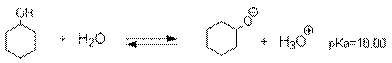
Алканолы являются слабыми основаниями
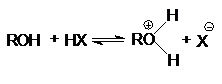
В присутствии сильных концентрированных кислот
равновесие в достаточной степени смещено в правую сторону.
Последняя реакция обусловливает легкое
отщепление уходящей группы, H2O, и это создает благоприятные условия
для реакции нуклеофильного замещения, например, при синтезе простых эфиров.
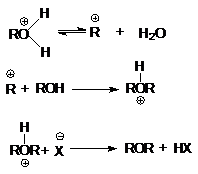
или суммарно: ROH + ROH ROH + H2O
при замещении гидроксильной группы на галоген или другой кислотный остаток
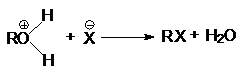
или суммарно:
ROH + HX ® RX + H2O
при алкилировании
аренов спиртами
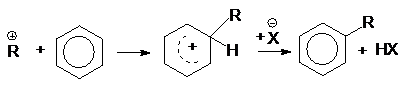
или суммарно:
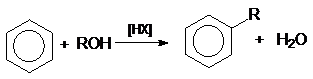
в реакции дегидратации спиртов
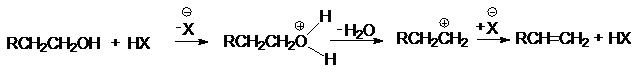
в различных перегруппировках,
связанных с перегруппировками карбкатиона
а) миграция алкильных групп
б) миграция водорода
в) аллильная
перегруппировка
г) ретропинаколиновая перегруппировка
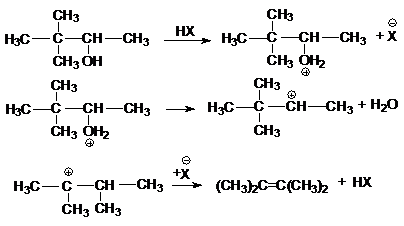
или суммарно:
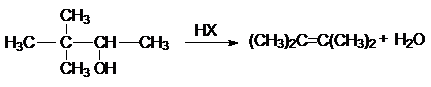
Нуклеофильные
свойства спиртов.
Нуклеофильные свойства спиртов проявляются при их
взаимодействии с галогенангидридами карбоновых и
неорганических кислот, которые обладают высокореакционноспособным
реакционным центром. Так, при замещении ОН-группы
на галоген тионилхлоридом:
ROH +
SOCl2 R¾Cl + SO2 + HCl
cпирт
успешно атакует положительно заряженный атом серы. Последующее отщепление
аниона хлора облегчается его протонированием
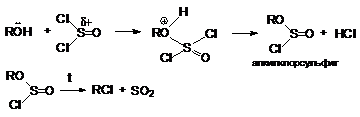
Реакция интересна тем, что она протекает с сохранением
конфигурации карбкатионного атома углерода.
Объясняется это тем, что в алкилхлорсульфите конфигурация та же что и в исходном спирте. При нагревании алкилхлорсульфит распадается на ионную пару.
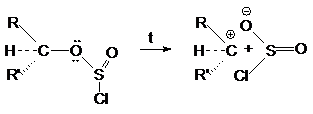
Поскольку анион SO2Cl может
атаковать карбкатион с той же стороны, то
конфигурация сохраняется. Механизм этих реакций обозначают символом SN i -внутримолекулярное
нуклеофильное замещение (substitution nucleofilic intramolecula).
Взаимодействие
с галогенидами фосфора.
3ROH + PBr3 ® 3RBr + H3PO3
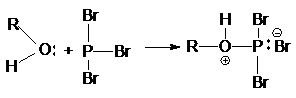
![]() RCOOH + R’OH
RCOOH + R’OH ![]() RCOOR’ + H2O
RCOOR’ + H2O
RCOCl + R’OH ® RCOOR’ + HCl
(RCO)2O + R’OH ® RCOOR’ +
RCOOH
![]() HONO2 + ROH RONO2 + H2O
HONO2 + ROH RONO2 + H2O
RCH2OH
+ 1/2 O2 RCHO + H2O
R2CHOH
+ 1/2 O2 R2C
= O + H2O
Аналогичный результат получают при
действии CrO3 в кислой среде
K2Cr2O7, KMnO4 в кислой и
нейтральных средах, MnO2 в кислой среде.
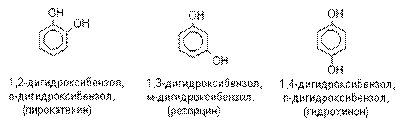
Многоатомные спирты. Диолы.
Многоатомные спирты – производные алканов,
в которых два или большее число атомов водорода при различных углеродных атомах
замещены на гидроксильную группу. Если многоатомные спирты содержат две гидроксильные
группы, то они называются диолами или гликолями. Если
они содержат три гидроксильные группы, то они называются триолами.
Общее название многоатомных спиртов – полиолы.
Наиболее важными из многоатомных спиртов являются диолы.
В зависимости от взаимного расположения
гидроксильных групп различают 1,2– диолы, 1,3–диолы, 1,4–диолы и т.д. У 1,2–диолов группы ОН расположены у двух соседних углеродных атомов, например,
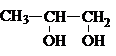
У 1,3–диолов
группы ОН расположены при атомах углерода, находящихся в 1,3–положении по
отношению друг к другу, например, HOCH2CH2CH2OH. У 1,4– диолов группы ОН
расположены при атомах углерода, находящихся в 1,4–положении по отношении друг
к другу. 1,2- ,1,3– и 1,4–диолы иногда называют a- ,b- и g- гликолями соответственно.
При образовании названий диолов используют номенклатуру IUPAC и
тривиальную номенклатуру. В системе IUPAC
пользуются общими правилами этой номенклатуры, в тривиальной номенклатуре диолы часто называют гликолями.
1. Гидролиз дигалогеналканов
или галогенгидринов
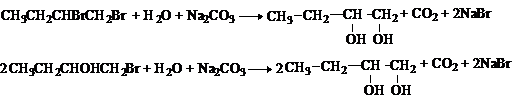
2. Гидратация циклических эфиров
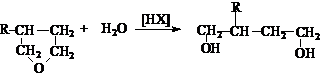
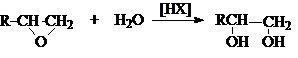
3. Окисление алкенов пероксидом
водорода или перманганатом калия

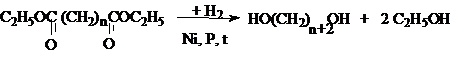
4. Каталитическое восстановление эфиров
дикарбоновых кислот.
Низшие члены ряда – густые жидкости,
высшие – кристаллические вещества. Температуры кипения гликолей выше, чем
температуры кипения спиртов близкой молекулярной массы. Низшие гликоли хорошо
растворимы в воде. Растворимость гликолей в воде выше, чем растворимость близких
им по молекулярной массе спиртов. Эти факты объясняются двумя причинами. Во–первых, наличие двух спиртовых функций обусловливает
большее число межмолекулярных водородных связей, образуемых молекулами
гликолей, во–вторых, эти связи прочнее, так как одна спиртовая группа действует
на другую как акцептор, увеличивая ее кислотность. Гликоли имеют сладковатый
вкус, плотность большинства из них больше 1.
Химические свойства
гликолей аналогичны свойствам одноатомных спиртов. Они могут вступать в химические
взаимодействия как одной, так и двумя гидроксильными группами.
1)
Кислотность
Гликоли обладают
более высокой кислотностью по сравнению с одноатомными спиртами. Поэтому они также, как и последние, легко вступают во взаимодействие со
щелочными металлами, и в отличие от одноатомных спиртов, реагируют со щелочами
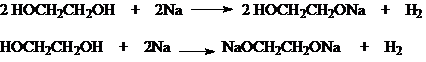
![]()
и некоторыми
основными гидроксидами
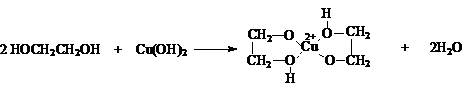
Последняя реакция
легко наблюдается как растворение Сu(OH)2
с образованием синего раствора. Эта реакция является качественной на многоатомные
спирты.
2) Образование полных и неполных эфиров с
неорганическими и органическими кислотами.
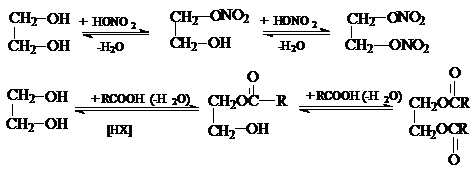
Если в качестве этерифицирующих агентов используются ангидриды и хлорангидриды карбоновых кислот, то реакции этерификации диолов протекают необратимо:
3) Окисление диолов
Процесс окисления диолов протекает обычно по сложной схеме, включающей последовательные
и параллельные реакции.
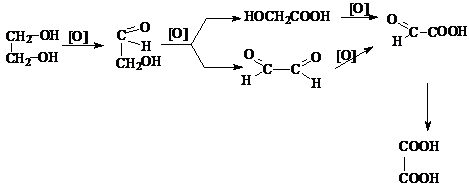
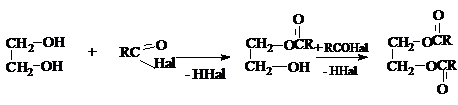
При этом, как и в
случае одноатомных спиртов, по продуктам окисления можно судить о структуре диола. Так, если при окислении диола
состава C4H8(OH)2 в продуктах окисления обнаруживается только
уксусная кислота, то диол может иметь только формулу
2,3 – бутандиола.
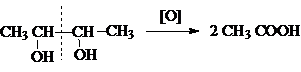
В этом плане
характерной реакцией окисления, являющейся тестом на установление структуры
1,2–диолов (a-гликолей), является реакция Малапрада,
в которой в качестве окислителей используется тетраацетат
свинца или периодат натрия.
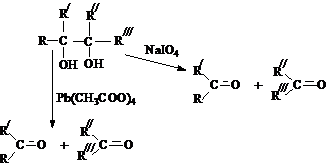
4)
Дегидратация
Реакции дегидратации диолов могут иметь межмолекулярный или внутримолекулярный
характер. Эти реакции относятся к реакциям нуклеофильного замещения. Поэтому
для электрофильного отщепления «плохой» уходящей
группы – OH требуется кислотный катализ.
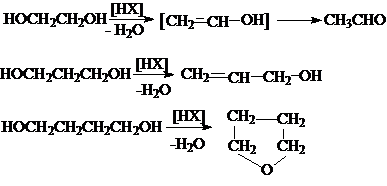
Своеобразным типом
дегидратации является пинаколиновая перегуппировка.
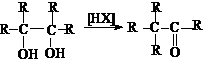
Пинаколиновая перегруппировка является типичной реакцией перегруппировки
карбкатионов, которые образуются при протонировании пинаконов.
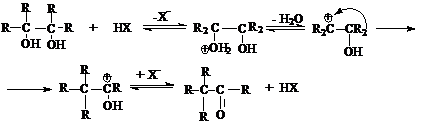
Фенолы.
Фенолы – гидроксилпроизводные
ароматических углеводородов, в которых группа ОН связана с атомом углерода
ароматического ядра.
По количеству ароматических ядер в молекуле различают
собственно фенолы, а также нафтолы, антролы, фенантролы и др. По числу гидроксильных групп различают
одно-, двух-, трех-, многоатомные фенолы.
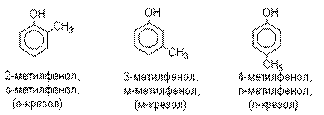
Названия фенолов образуют от названий
соответствующих аренов с добавлением суффикса –ол. Простейший
фенол – гидроксибензол С6Н5ОН называют
просто фенол. При наличии нескольких
заместителей начало нумерации определяет гидроксильная группа и эти соединения
рассматриваются как производные фенола. Иногда в соединениях сложного строения
наличие гидроксильной группы обозначают префиксом гидрокси-. Многие фенолы имеют тривиальные названия.
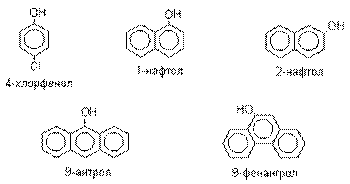
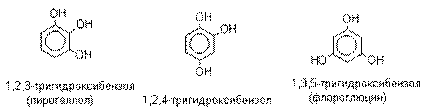
Фенол выделяют из каменноугольной смолы.
Существует многочисленные синтетические методы получения фенола, промышленные и
лабораторные.
1)
Замещение сульфогруппы на
гидроксил
![]()
![]()
2).Замещение
галогена на гидроксил
Арилгалогениды, не содержащие активирующих
электроноакцепторных заместителей, вступают в реакцию обмена в очень жестких
условиях. Фенол получают нагреванием хлорбензола с 15-20%-ным водным раствором гидроксида натрия при 360-390оС и давлении
280-300 атм.
C6H5Cl + 2NaOH C6H5O-Na+
+ NaCl + H2O
C6H5O-Na+ + HCl ® C6H5OH + NaCl
В этих условиях реакция идет по ариновому механизму.
2)
Замещение диазогруппы на
гидроксил
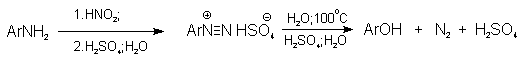
Метод включает диазотирование
первичного амина с последующими разложением соли диазония в водном растворе серной кислоты. Замещение диазогруппы на гидроксил протекает по SN1-механизму.
Так как промежуточно образующийся
арилкатион может реагировать с любым нуклеофилом, имеющимся в реакционной среде, наиболее
целесообразно использовать для гидролиза в фенолы гидросульфаты диазония. В качестве побочного продукта при этом образуется
эфир ArOSO3H, который легко гидролизуется
в фенол.
4)
Получение фенола из гидропероксида кумола
Современный промышленный метод получения
фенола заключается в кислотно-катализируемом разложении гидропероксида
кумола. Исходное вещество для всего цикла превращений - кумол получают алкилированием бензола пропиленом по Фриделю-Крафтсу.
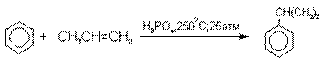
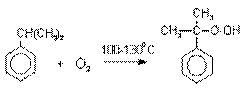
Далее кумол окисляют кислородом воздуха
при 100-130оС до гидропероксида кумола.
Разложение гидропероксида
кумола до фенола и ацетона проводят в присутствии 1% водной серной кислоты при
50-90оС.
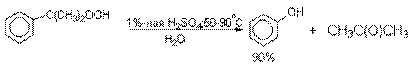
В целом это очень экономичный способ
получения одновременно двух важнейших продуктов - ацетона и фенола.
Физические
свойства и строение.
Фенолы – жидкие или кристаллические
вещества с сильным характерным запахом. При хранении на воздухе быстро темнеют
из-за окисления. Ограниченно растворимы в воде.
Гидроксигруппа и ароматическое кольцо образуют сопряженную систему. Гидроксигруппа проявляет электронодонорные свойства за счет +М-эффекта, который превышает по силе –I-эффект.
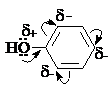
Фенол и его гомологи – полярные соединения. Дипольный
момент направлен в сторону бензольного кольца.
Взаимодействии неподеленной пары электронов кислорода
с p-системой кольца
обусловливает электронодонорные свойства этих
соединений. Энергия ионизации фенола составляет 8,5-8,6 эВ, что ниже, чем у
бензола и спиртов.
Для фенолов характерны нуклеофильные
свойства, которые усиливаются при превращении их в феноксид-анионы
в результате ионизации полярной связи О-Н. При этом электрофилы могут реагировать как по атому кислорода, так и
по атомам углерода ароматического кольца фенолов или феноксид-анионов.
Фенолы являлются
слабыми ОН-кислотами, но
значительно превосходят по кислотности спирты. Например, фенол в 108 раз
более сильная ОН-кислота по
сравнению с циклогексанолом.
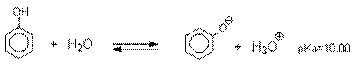
Причина более высокой кислотности фенолов состоит в стабилизации феноксид-аниона за счет делокализации отрицательного заряда с участием ароматического кольца.
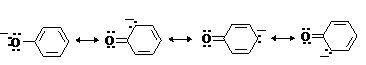
Влияние заместителя в бензольном кольце на кислотность фенолов согласуется с
представлениями об их электронных эффектах. Электроноакцепторные заместители,
особенно в орто- и пара-положениях,
усиливают, а электронодонорные - понижают кислотные
свойства фенолов.
Величины рКа орто-,
мета- и пара-замещенных фенолов в воде при 25оС
|
Заместитель |
орто |
мета |
пара |
Заместитель |
орто |
мета |
пара |
|
H |
10.00 |
10.00 |
10.00 |
F |
8.73 |
9.29 |
9.89 |
|
CH3 |
10.29 |
10.09 |
10.26 |
Cl |
8.56 |
9.12 |
9.41 |
|
C(CH3)3 |
10.62 |
10.12 |
10.23 |
Br |
8.45 |
9.03 |
9.37 |
|
C6H5 |
10.01 |
9.64 |
9.55 |
I |
8.51 |
9.03 |
9.33 |
|
OCH3 |
9.98 |
9.65 |
10.21 |
NO2 |
7.23 |
8.36 |
7.15 |
Большинство фенолов легко растворяется в
водных растворах щелочей МОН с образованием фенолятов ArOM.
Получение
простых и сложных эфиров фенолов.
Нуклеофильные свойства атома кислорода фенолов
понижены по сравнению со спиртами в результате сопряжения. Как следствие этого ариловые эфиры карбоновых кислот нельзя получать прямой
этерификацией фенолов карбоновыми кислотами. Сложные эфиры получают ацилированием фенолов или их Na-и K-солей галогенангидридами или ангидридами кислот.
ArOH + RCOX ® RCOOAr + HX
ArO-Na+ +
RCOX ® RCOOAr + NaX
(X=Cl, OCOR)
Феноляты легко алкилируются,
алкилгалогенидами и диалкилсульфатами
с образованием простых эфиров.
ArO-Na+ + RX
® ArOR + NaX
(X=Hal,
ROSO3)
Метиловые эфиры фенолов получают также
действием диазометана в эфирном растворе.
ArOH + CH2N2 ® AROCH3
+ N2
В отличие от спиртов фенолы как более
сильные кислоты метилируются диазометаном
в отсутствие катализатора.
Перегруппировка Кляйзена аллилариловых эфиров.
Аллиловый эфир фенола при нагревании до 200-220оС
превращается в орто-аллилфенол,
т.е. аллильная группа мигрирует в орто-положение бензольного кольца.
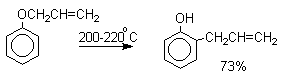
Если оба орто-положения заняты
заместителями, то аллильная группа перемещается в пара-положение:
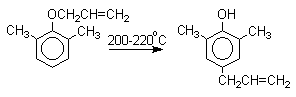
Реакции электрофильного замещения в ароматическом кольце.
Гидроксильная группа относится к числу
групп, активирующих электрофильное замещение в
ароматическом кольце и направляющих заместитель в орто- и пара-положения.
Фенолы вступают практически во все типичные реакции электрофильного
замещения как с сильными, так и со слабыми электрофильными агентами.
Галогенирование фенолов не требует
катализа кислотами Льюиса и легко осуществляется под действием молекулярного
галогена. Галогенирование фенола молекулярным бромом или хлором в полярной
среде практически невозможно остановить на стадии моногалогенирования,
поскольку реагирующей частицей здесь является фенолят-ион, который содержит очень
сильную активирующую группу –O-. Скорость галогенирования фенолят-иона по крайней мере в тысячу раз выше, чем
фенола. Галогензамещенный фенол является более
сильной кислотой, чем сам фенол, что облегчает введение второго и третьего
атома галогена в орто- и пара-положения.
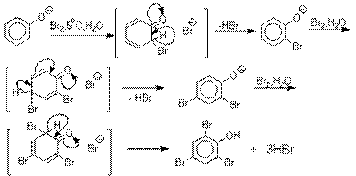
Бромирование фенола в воде приводит к образованию нерастворимого
2,4,6-трибромфенола. Эта реакция настолько чувствительна, что позволяет обнаружить
фенол в концентрации 10-5М в водном растворе. 2,4,6-Трибромфенол
взаимодействует еще с одним молем брома с образованием
2,4,4,6-тетрабромциклогекса-2,5-диенона, окрашенного в желтый цвет.

При обработке продукта этой реакции
раствором гидросульфита натрия или другого слабого восстановителя он легко
превращается в исходный 2,4,6-трибромфенол.
При бромировании фенола в
растворе бромистоводородной кислоты диссоциация
полностью подавляется и галогенированию подвергается сам фенол. При этом в
зависимости от условий и количества галогена может быть получен
п-бромфенол или 2,4-дибромфенол.
Аналогичным образом протекает
хлорирование фенола хлором в соляной кислоте, но здесь получается значительное
количество о-хлорфенола. Моногалогензамещенные
производные фенолов удобно получать при галогенировании в неполярной среде, что
также исключает диссоциацию фенолов.
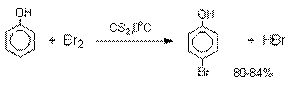
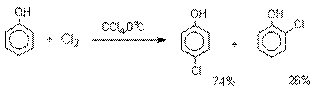
В качестве галогенирующего
агента кроме самих галогенов можно использовать комплексы галогенов с диоксаном, ДМФА.
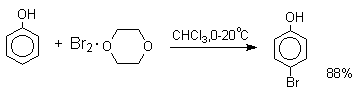
Во всех случаях соотношение пара/орто-изомеров при бромировании и иодировании
значительно выше, чем при хлорировании.
Нитрование фенолов разбавленной
20-25%-ной азотной кислотой приводит к получению смеси орто- и пара-нитрофенолов.
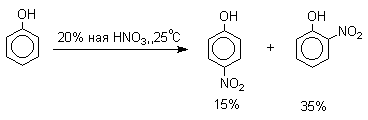
Даже в таких мягких условиях нитрование
сопровождается окислением фенола и этот процесс становится доминирующим, если
для нитрования использовать концентрированную азотную кислоту. Поэтому для
получения 2,4,6-тринитрофенола (пикриновой кислоты) используют видоизмененный способ
нитрования. Фенол первоначально сульфируют до
4-гидрокси-1,3-бензолдисульфокислоты, а затем нитруют азотной кислотой.
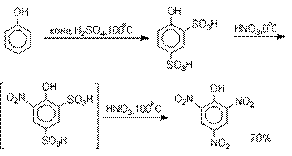
Вторая стадия по существу представляет
собой электрофильное ипсо-замещение
сульфогруппы на нитрогруппу.
Сульфирование.
Моносульфирование фенола серной кислотой приводит к образованию смеси орто- и пара-изомеров гидроксибензолсульфоксилоты. При 20оС в реакционной
смеси содержится 49% орто-изомера
и 51% пара-изомера,
тогда как при 120оС доля пара-изомера возрастает до 96%.
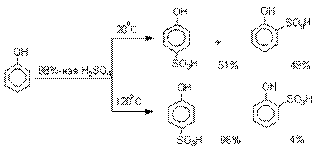
Изменение в соотношении продуктов
сульфирования обусловлено обратимостью реакций, когда в равновесии преобладает термодинамически более стабильный пара-изомер.
Сульфирование в орто-положение
протекает с большей скоростью, но орто-гидроксибензолсульфокислота легко гидролизуется
на исходные реагенты в отличие от пара-изомера, для которого скорость гидролиза мала.
Нитрозирование фенолов осуществляется с помощью азотистой кислоты в
воде или уксусной кислоте. Эта реакция отличается очень высокой региоселективностью и замещение
идет в пара-положение
к гидроксильной группе. Типичное распределение орто- и пара-изомеров
при нитрозировании можно проиллюстрировать на примере
самого фенола.
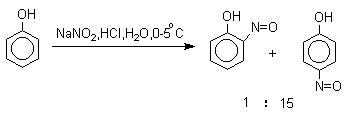
Алкилирование и ацилирование по Фриделю-Крафтсу.
Так как фенолы взаимодействуют с
галогенидами алюминия и другими кислотами Льюиса с образованием солей типа
ArOAlCl2, прямое их алкилирование
в условиях реакции Фриделя-Крафтса провести не
удается. Фенолы алкилируют алкенами
и спиртами в условиях кислотного катализа. В качестве катализаторов
предпочитают использовать серную, фтористоводородную, фосфорную кислоты или
катиониты КУ-2 и другие катионообменные смолы. Таким
образом, из крезола и изобутилена в промышленности получают пространственно
затрудненный фенол - 2,6-ди-трет-бутил-4-метилфенол
(ионол), который широко применяется для стабилизации
полимеров.
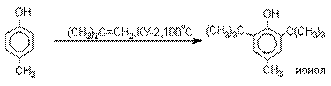
Аналогично из фенола и изопропилового
спирта получается 2,4,6-триизопропилфенол.
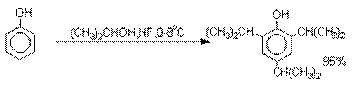
Ацилирование фенолов в классических условиях реакции Фриделя-Крафтса комплексом ацилгалогенида
и хлорида алюминия приводит к неудовлетворительным результатам, так как ацилированию подвергается гидроксильная группа фенола.
Более эффективна такая модификация этого метода, когда в качестве ацилирующего агента используется комплекс карбоновой кислоты
и трехфтористого бора. Ацильная группа при этом
вводится практически исключительно в пара-положение бензольного кольца. Так, например, фенол при взаимодействии
с комплексом уксусной кислоты и BF3 дает пара-гидроксиацетофенон с 95%-ным
выходом.
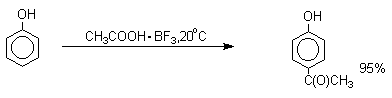
Наиболее общий метод получения гидроксикетонов ароматического ряда основан на перегруппировке Фриса.
Ариловые эфиры карбоновых кислот при нагревании с
AlCl3 или AlBr3 перегруппировываются в изомерные орто- и пара-гидроксикетоны.
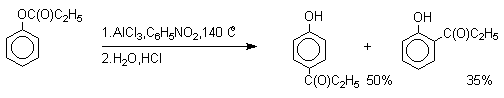
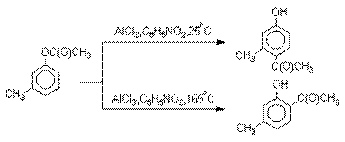
Соотношение орто- и пара-изомеров
зависит главным образом от температуры и растворителя. В более жестких условиях
преобладает орто-гидроксикетон,
а при 20-25оС - пара-гидроксикетон.
Механизм перегруппировки Фриса, по-видимому, заключается в межмолекулярном ацилировании орто- или пара-положения бензольного кольца арилового
эфира комплексом второй молекулы сложного эфира и AlCl3 с образованием
ацильного производного гидроксикетона
и фенола.
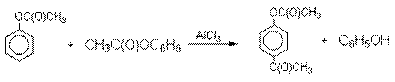
Перегруппировка завершается
межмолекулярным переносом ацильной группы к фенолу.
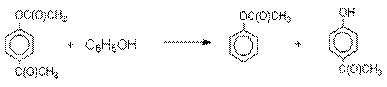
Формилирование.
Формилирование – это введение группы СНО (см. лек.35). Синтетически
наиболее важными являются формилирование фенолов по Вильсмейеру-Хааку и Реймеру-Тиману.
Реакция Вильсмейера-Хаака.
N-Алкиламиды муравьиной кислоты - N,N-диметилформамид (ДМФА) и N-метилформамид
в присутствии хлорокиси фосфора являются региоселективными формилирующими
агентами. С помощью этих реагентов альдегидная группа вводится в пара-положение по
отношению к ОН -группе. Эту реакцию можно также
рассматривать как ацилирование, где роль катализатора
(кислоты Льюиса) выполняют хлорокись фосфора (POCl3).
Наиболее эффективна система ДМФА-POCl3, в которой ДМФА служит и
реагентом, и растворителем. Электрофильным агентом в
реакции Вильсмейера-Хаака является иминиевая соль, которая образуется при взаимодействии ДМФА
и хлорокиси фосфора.
Иминиевая соль при необходимости может быть выделена в индивидуальном
виде, однако обычно ее не выделяют и используют непосредственно после ее образования.
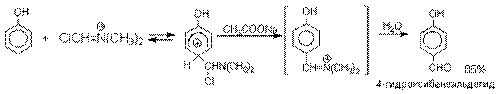
Реакция Вильсмейера-Хаака
чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие
выходы ароматических гидроксиальдегидов.
Реакция Реймера-Тимана.
Формилирование фенолов по Реймеру-Тиману
достигается при нагревании смеси фенола и большого избытка хлороформа с водным
раствором гидроксида натрия при 50-70оС.
Выходы альдегидов обычно невелики и редко превышают 30%, однако метод исключительно
прост и доступен в практическом отношении. Главное достоинство реакции Реймера-Тимана заключается в преимущественном образовании орто-, а не пара-изомеров, как это имеет место в реакции Вильсмейера-Хаака.
Механизм включает образование дихлоркарбена как интермедиата. Дихлоркарбен: CCl2 выполняет
роль электрофильного агента по отношению к феноксид-иону, образующемуся в щелочной среде.
Предполагаемый механизм реакции Реймера-Тимана может
быть представлен следующей последовательностью превращений:
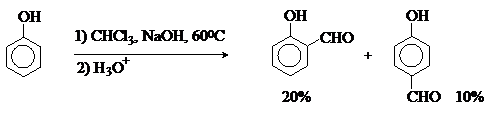
Реакция протекает только в сильно
щелочной среде при наличии фенольного гидроксила, тогда как простые
эфиры фенолов и диалкиланилины не формилируются
в этих условиях.
Карбоксилирование феноксид-ионов (реакция Кольбе).
Будучи сильными нуклеофилами
феноксид-анионы способны
взаимодействовать с таким слабым электрофильным
реагентом как оксид углерода (IV). При
нагревании сухих фенолятов натрия или лития с СО2
при повышенном давлении, образуются натриевые или литиевые соли салициловой
кислоты.
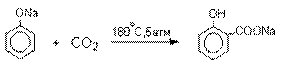
В аналогичных условиях из фенолятов
калия, рубидия и цезия получаются только соли пара-гидроксибензойной кислоты.
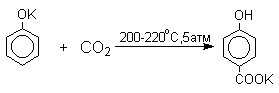
Такое различие в направлении карбоксилирования Na- и К-солей
фенола принято объяснять различием в хелатообразовании
этих двух катионов с атомом кислорода CO2 в переходном состоянии
реакции, приводящем к салициловой кислоте. Катионы натрия и, особенно, лития
значительно более эффективны по сравнению с катионом калия в способности к
образованию координационной связи с атомом кислорода.
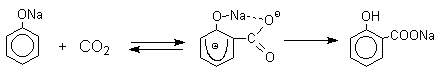
Предполагается, что для фенолятов калия,
рубидия и цезия электрофильная атака осуществляется
исключительно в пара-положение без какой-либо координации катиона по
атому кислорода.
В отличие от одноатомных фенолов
двухатомные и трехатомные фенолы карбоксилируются в
более мягких условиях. Так, резорцин карбоксилируется
при пропускании СО2 в водный раствор его дикалиевой соли при 50оС. При этом образуется
2,4-дигидроксибензойная кислота.
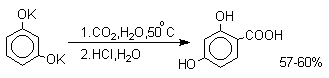
Конденсация с альдегидами и кетонами.
Фенолы реагируют с формальдегидом в
водном растворе в присутствии основания с образованием полимерного продукта,
получившего название феноло-формальдегидной смолы, карболита или бакелита.
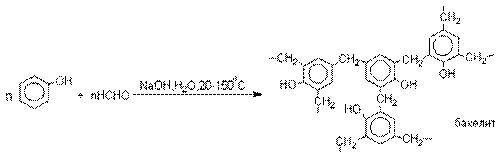
Взаимодействие феноксид-иона
с формальдегидом напоминает альдольную конденсацию с
той лишь разницей, что роль нуклеофильного агента вместо енолят-иона
выполняет амбидентный феноксид-ион,
а карбонильной компонентой является формальдегид.
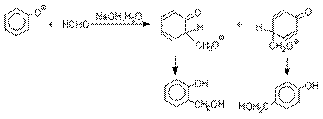
Подобно альдолям,
орто- и пара-изомеры
гидроксиметилфенола подвергаются дегидратации с
образованием хинонметидов – соединений, родственных орто- и пара-хинонам.
Окисление фенолов относится к числу сложных, многостадийных процессов, приводящих к продуктам разного строения. Механизм окисления может сильно меняться в зависимости от строения фенола и природы одно- или двухэлектронного окислителя. Механизм окисления включает стадию образования стабилизированных резонансом ароксильных радикалов.

Направление дальнейших превращений
зависит от характера заместителей в ароматическом кольце. При окислении
большинства фенолов образуется несколько различных форм димеров
в результате образования новых связей С-С между орто-орто, орто-пара- и пара-пара-положениями
ароксильных радикалов, а также новых С-О связей между
атомом кислорода одного радикала и орто- или пара-положением другой радикальной частицы. Всего, таким образом,
образуется потенциально не менее пяти различных типов димеров,
которые находится в равновесии с исходным ароксильным
радикалом. Например, для монозамещенного фенола:
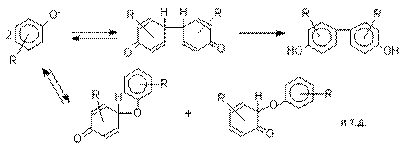
Димеры, возникающие в результате рекомбинации радикалов с
образованием новой С-С связи, называются хинолидами. Хинолиды далее изомеризуются с образованием изомерных
дигидроксибифенилов. Другой тип димеров,
содержащих центральную связь С-О, носит название хиноловых эфиров.
Ароксильные радикалы пространственно затрудненных фенолов, содержащие
в обоих орто- и пара-положении
третичные алкильные группы, мономерны и не проявляют
тенденции к образованию димеров в растворе. Например,
при окислении 2,4,6-три-трет-бутилфенола
гексацианоферратом(III) калия K3Fe(CN)6
в бинарной системе бензол-вода в инертной атмосфере образуется устойчивый
радикал одновалентного кислорода - три-трет-бутилфеноксил, окрашенный в синий цвет.
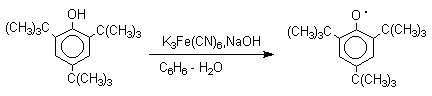
Этот радикал находится в мономерной
форме в 0,1 молярном растворе в бензоле или эфире, а также в кристаллическом
состоянии. Он очень чувствителен к действию кислорода воздуха, оксида азота
(IY), оксида азота (II) и других радикальных частиц. Стабильность ароксильных радикалов пространственно затрудненых
фенолов обусловливает их антиокислительные свойства.
Они выполняют роль ловушек свободных радикалов в
процессах пероксидного окисления. Активный свободный
радикал, ведущий цепь окисления, быстро взаимодействует с таким фенолом, давая
устойчивый ароксильный радикал, что приводит к обрыву
цепи.
ArOH + RO2s ® ArOs + ROOH
Пространственно затрудненные фенолы (ионол, гальваноксил) используют
как антиоксиданты, стабилизирующие синтетические каучуки, пищевые жиры, витамины
и др.