Лекция
№29-30
ФЕНОЛЫ
План
- Классификация и номенклатура.
- Методы получения.
- Физические свойства и строение.
- Химические свойства.
Фенолы
– гидроксилпроизводные ароматических углеводородов, в которых группа ОН связана
с атомом углерода ароматического ядра.
1. Классификация и номенклатура
По количеству ароматических ядер в молекуле различают собственно фенолы, а также нафтолы, антролы, фенантролы и др. По числу гидроксильных групп различают одно-, двух-, трех-, многоатомные фенолы.
Названия фенолов образуют от названий соответствующих аренов с добавлением суффикса –ол. Простейший фенол – гидроксибензол С6Н5ОН называют просто фенол. При наличии нескольких заместителей начало нумерации определяет гидроксильная группа и эти соединения рассматриваются как производные фенола. Иногда в соединениях сложного строения наличие гидроксильной группы обозначают префиксом гидрокси-. Многие фенолы имеют тривиальные названия.
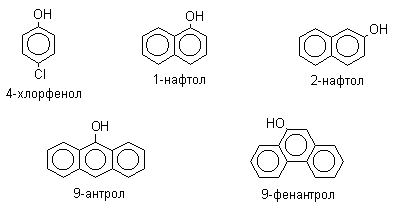
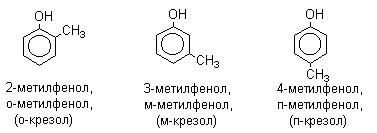
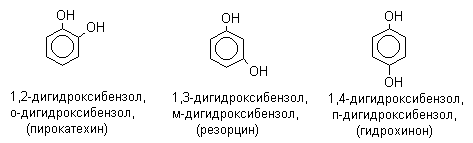
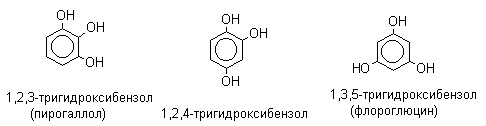
2. Методы получения
Фенол выделяют из каменноугольной смолы. Существует многочисленные синтетические методы получения фенола, промышленные и лабораторные.
1) Замещение сульфогруппы на гидроксил
![]()
![]()
Наиболее старый промышленный метод получения фенолов заключается в
сплавлении щелочных арилсульфонатов с твердым гидроксидом натрия или калия или
со сравнительно легкоплавкой смесью этих гидроксидов при 300-350оС.
Хотя точный механизм реакции в расплаве двух ионных соединений неизвестен, ее
следует отнести к процессам нуклеофильного ароматического замещения, где
гидроксид-ион играет роль нуклеофильного агента, а сульфит-ион - уходящей
группы.
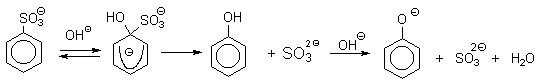
Для получения самого фенола метод щелочного плавления в настоящее время не используется, но он широко используется для получения 2-нафтола, резорцина, ализарина и других фенолов.
2).Замещение галогена на гидроксил
Арилгалогениды, не содержащие активирующих электроноакцепторных заместителей, вступают в реакцию обмена в очень жестких условиях. Фенол получают нагреванием хлорбензола с 15-20%-ным водным раствором гидроксида натрия при 360-390оС и давлении 280-300 атм.
C6H5Cl
+ 2NaOH ® C6H5O-Na+ + NaCl + H2O
C6H5O-Na+ + HCl ® C6H5OH + NaCl
В
этих условиях реакция идет по ариновому механизму (см. лек.№24).
В присутствии солей меди (II), играющих роль катализатора, ариновый механизм полностью подавляется из-за резкого ускорения прямого замещения галогена по SNAr-механизму. Применение солей меди (II) позволяет проводить региоселективное замещение галогена на гидроксил без примеси какого-либо другого изомерного фенола.
![]()
Введение в молекулу арилгалогенида электроноакцепторных заместителей в орто- или пара-положения увеличивают скорость обмена галогена на гидроксил в некатализируемой реакции. Для таких реакций предложен SNAr-механизм замещения.
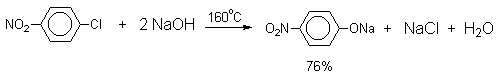
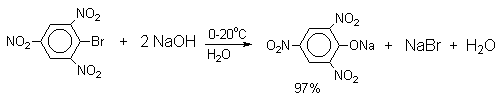
3) Замещение диазогруппы на гидроксил
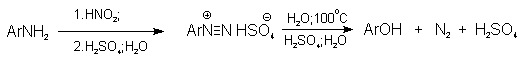
Метод
включает диазотирование первичного амина с последующими разложением соли
диазония в водном растворе серной кислоты. Замещение диазогруппы на гидроксил
протекает по SN1-механизму. Так как промежуточно образующийся
арилкатион может реагировать с любым нуклеофилом, имеющимся в реакционной среде,
наиболее целесообразно использовать для гидролиза в фенолы гидросульфаты
диазония. В качестве побочного продукта при этом образуется эфир ArOSO3H,
который легко гидролизуется в фенол. См. также лек. №24 и 43.
4) Получение фенола из гидропероксида кумола
Современный промышленный метод получения фенола заключается в кислотно-катализируемом разложении гидропероксида кумола. Исходное вещество для всего цикла превращений - кумол получают алкилированием бензола пропиленом по Фриделю-Крафтсу.
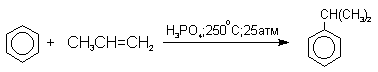
Далее кумол окисляют кислородом воздуха при 100-130оС до гидропероксида кумола.
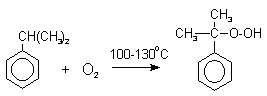
Эта реакция протекает по цепному радикальному механизму с участием кумильного радикала.
![]()
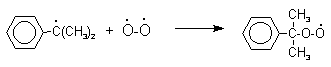
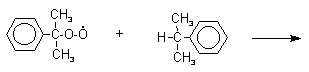
![]()
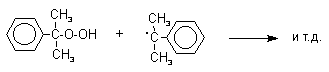
Разложение гидропероксида кумола до фенола и ацетона проводят в присутствии 1% водной серной кислоты при 50-90оС.
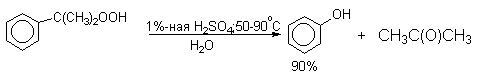
Процесс разложения гидропероксида по своему механизму напоминает перегруппировки карбкатионов. Различие заключается в том, что миграция фенила происходит к положительно заряженному атому кислорода.
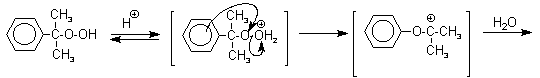
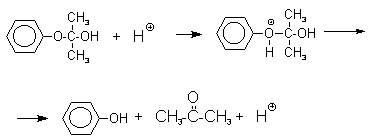
В целом это очень экономичный способ получения одновременно двух важнейших продуктов - ацетона и фенола.
3. Физические свойства и строение
Фенолы – жидкие или кристаллические вещества с сильным характерным запахом. При хранении на воздухе быстро темнеют из-за окисления. Ограниченно растворимы в воде.
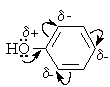
Гидроксигруппа и ароматическое кольцо образуют сопряженную систему. Гидроксигруппа проявляет электронодонорные свойства за счет +М-эффекта, который превышает по силе –I-эффект.
Фенол и его гомологи – полярные соединения. Дипольный момент направлен в сторону бензольного кольца. Взаимодействии неподеленной пары электронов кислорода с p -системой кольца обусловливает электронодонорные свойства этих соединений. Энергия ионизации фенола составляет 8,5-8,6 эВ, что ниже, чем у бензола и спиртов.
Спектральные характеристики.
Введение гидроксильной группы в бензольное кольцо вызывает сдвиг полос поглощения в УФ-спектрах в длинноволновую область и усиливает их: 210 (e 6200) и 270 (e 1450) нм.
В ИК-спектрах характеристические полосы поглощения валентных колебаний группы ОН лежат в области 3390-3600 см-1 и зависят от растворителя и концентрации.
В спектрах ПМР сигнал протонов группы ОН находится в широком диапазоне и зависит от температуры, концентрации, рН и растворителя (в дейтерохлороформе d 4,5-7,5 м.д.)
4. Химические свойства
Для фенолов характерны нуклеофильные свойства, которые усиливаются при превращении их в феноксид-анионы в результате ионизации полярной связи О-Н. При этом электрофилы могут реагировать как по атому кислорода, так и по атомам углерода ароматического кольца фенолов или феноксид-анионов.
Кислотные свойства
Фенолы являлются слабыми ОН-кислотами, но значительно превосходят по кислотности спирты. Например, фенол в 108 раз более сильная ОН-кислота по сравнению с циклогексанолом.
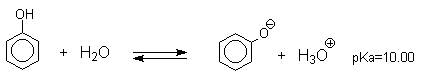
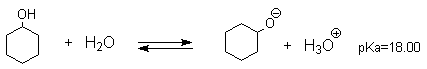
Причина более высокой кислотности фенолов состоит в стабилизации феноксид-аниона за счет делокализации отрицательного заряда с участием ароматического кольца.
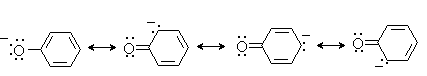
Влияние заместителя в бензольном кольце на кислотность фенолов согласуется с представлениями об их электронных эффектах. Электроноакцепторные заместители, особенно в орто- и пара-положениях, усиливают, а электронодонорные - понижают кислотные свойства фенолов.
Величины рКа орто-, мета- и пара-замещенных фенолов в воде при 25оС
|
Заместитель |
орто |
мета |
пара |
Заместитель |
орто |
мета |
пара |
|
H |
10.00 |
10.00 |
10.00 |
F |
8.73 |
9.29 |
9.89 |
|
CH3 |
10.29 |
10.09 |
10.26 |
Cl |
8.56 |
9.12 |
9.41 |
|
C(CH3)3 |
10.62 |
10.12 |
10.23 |
Br |
8.45 |
9.03 |
9.37 |
|
C6H5 |
10.01 |
9.64 |
9.55 |
I |
8.51 |
9.03 |
9.33 |
|
OCH3 |
9.98 |
9.65 |
10.21 |
NO2 |
7.23 |
8.36 |
7.15 |
Большинство фенолов легко растворяется в водных растворах щелочей МОН с образованием фенолятов ArOM.
Получение простых и сложных эфиров фенолов
Нуклеофильные свойства атома кислорода фенолов понижены по сравнению со спиртами в результате сопряжения. Как следствие этого ариловые эфиры карбоновых кислот нельзя получать прямой этерификацией фенолов карбоновыми кислотами. Сложные эфиры получают ацилированием фенолов или их Na-и K-солей галогенангидридами или ангидридами кислот.
ArOH + RCOX ® RCOOAr + HX
ArO-Na+ + RCOX ® RCOOAr + NaX
(X=Cl, OCOR)
Феноляты легко алкилируются , алкилгалогенидами и диалкилсульфатами с образованием простых эфиров.
ArO-Na+ + RX ® ArOR + NaX
(X=Hal, ROSO3)
Метиловые эфиры фенолов получают также действием диазометана в эфирном растворе.
ArOH + CH2N2 ® AROCH3 + N2
В отличие от спиртов фенолы как более сильные кислоты метилируются диазометаном в отсутствие катализатора.
Перегруппировка Кляйзена аллилариловых эфиров
Аллиловый эфир фенола при нагревании до 200-220оС превращается в орто-аллилфенол, т.е. аллильная группа мигрирует в орто-положение бензольного кольца.
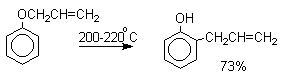
Если оба орто-положения заняты заместителями, то аллильная группа перемещается в пара-положение:
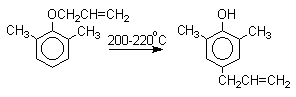
Установлено, что и орто- и пара-перегруппировки являются внутримолекулярными реакциями первого порядка. Миграция в орто-положение сопровождается инверсией аллильной группы, т.е. она присоединяется к бензольному кольцу g -углеродным атомом.
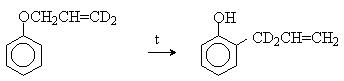
Из этого следует, что переходное состояние перегруппировки Кляйзена должно быть циклическим шестизвенным. Такое переходное состояние включает шесть p -электронов и является ароматическим, что составляет движущую силу этой термической перегруппировки. На последней стадии происходит изомеризация циклогексадиенона в о-аллилфенол. Эта стадия аналогична изомеризации кетона в енольную форму.
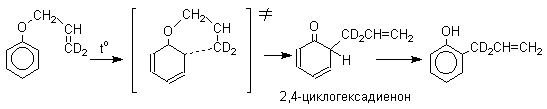
2,4-Циклогексадиенон является интермедиатом перегруппировки аллилариловых эфиров. Такой интермедиат может быть выделен при перегруппировке аллилового эфира 2,6-диметилфенола, когда аллильная группа мигрирует в пара-положение, поскольку енолизация кетона в фенол в этом случае не может происходить из орто-положения. Конечным результатом двух последовательных миграций аллильной группы является сохранение структуры мигрирующей группы.
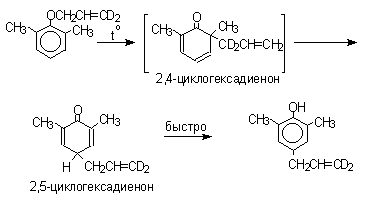
Кроме того, при проведении перегруппировки в присутствии малеинового ангидрида 2,4-циклогексадиенон улавливается в виде аддукта диенового синтеза.
Реакции электрофильного замещения в ароматическом кольце
Гидроксильная группа относится к числу групп, активирующих электрофильное замещение в ароматическом кольце и направляющих заместитель в орто- и пара-положения. Фенолы вступают практически во все типичные реакции электрофильного замещения как с сильными, так и со слабыми электрофильными агентами.
Галогенирование
Галогенирование фенолов не требует катализа кислотами Льюиса и легко осуществляется под действием молекулярного галогена. Галогенирование фенола молекулярным бромом или хлором в полярной среде практически невозможно остановить на стадии моногалогенирования, поскольку реагирующей частицей здесь является фенолят-ион, который содержит очень сильную активирующую группу –O-. Скорость галогенирования фенолят-иона по крайней мере в тысячу раз выше, чем фенола. Галогензамещенный фенол является более сильной кислотой, чем сам фенол, что облегчает введение второго и третьего атома галогена в орто- и пара-положения.
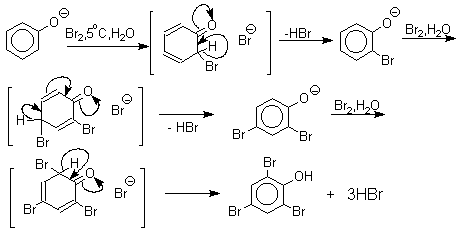
Бромирование фенола в воде приводит к образованию нерастворимого 2,4,6-трибромфенола. Эта реакция настолько чувствительна, что позволяет обнаружить фенол в концентрации 10-5М в водном растворе. 2,4,6-Трибромфенол взаимодействует еще с одним молем брома с образованием 2,4,4,6-тетрабромциклогекса-2,5-диенона, окрашенного в желтый цвет.
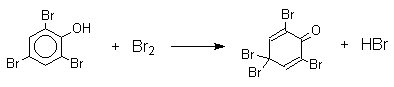
При обработке продукта этой реакции раствором гидросульфита натрия или
другого слабого восстановителя он легко превращается в исходный
2,4,6-трибромфенол.
При бромировании фенола в растворе бромистоводородной кислоты диссоциация
полностью подавляется и галогенированию подвергается сам фенол. При этом в
зависимости от условий и количества галогена может быть получен п-бромфенол или
2,4-дибромфенол.
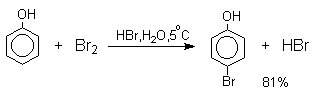
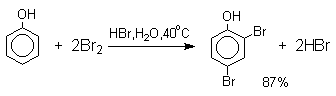
Аналогичным образом протекает хлорирование фенола хлором в соляной кислоте, но здесь получается значительное количество о-хлорфенола. Моногалогензамещенные производные фенолов удобно получать при галогенировании в неполярной среде, что также исключает диссоциацию фенолов.
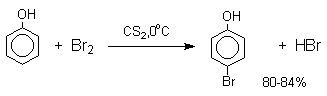
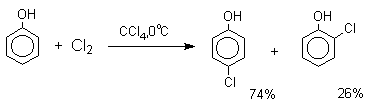
В качестве галогенирующего агента кроме самих галогенов можно использовать комплексы галогенов с диоксаном, ДМФА.
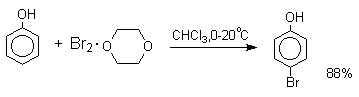
Во всех случаях соотношение пара/орто-изомеров при бромировании и иодировании значительно выше, чем при хлорировании.
Нитрование
Нитрование фенолов разбавленной 20-25%-ной азотной кислотой приводит к получению смеси орто- и пара-нитрофенолов.
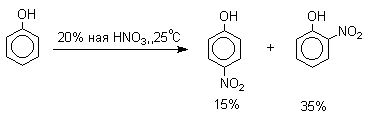
Даже в таких мягких условиях нитрование сопровождается окислением фенола и этот процесс становится доминирующим, если для нитрования использовать концентрированную азотную кислоту. Поэтому для получения 2,4,6-тринитрофенола (пикриновой кислоты) используют видоизмененный способ нитрования. Фенол первоначально сульфируют до 4-гидрокси-1,3-бензолдисульфокислоты, а затем нитруют азотной кислотой.
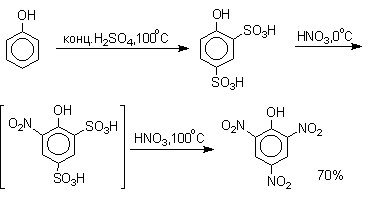
Вторая стадия по существу представляет собой электрофильное ипсо-замещение
сульфогруппы на нитрогруппу.
Для нитрования фенолов в качестве нитрующего агента кроме азотной кислоты можно
использовать ацетилнитрат и N2O4. Эти реагенты
способствуют преимущественному нитрованию в орто-положение к
гидроксильной группе.
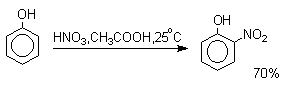
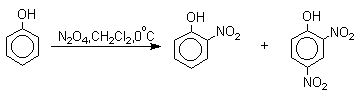
Сульфирование
Моносульфирование фенола серной кислотой приводит к образованию смеси орто- и пара-изомеров гидроксибензолсульфоксилоты. При 20оС в реакционной смеси содержится 49% орто-изомера и 51% пара-изомера, тогда как при 120оС доля пара-изомера возрастает до 96%.
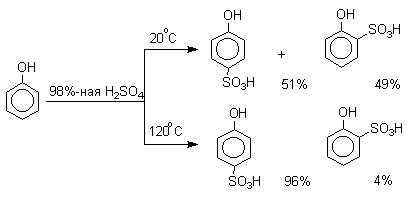
Изменение в соотношении продуктов сульфирования обусловлено обратимостью реакций, когда в равновесии преобладает термодинамически более стабильный пара-изомер. Сульфирование в орто-положение протекает с большей скоростью, но орто-гидроксибензолсульфокислота легко гидролизуется на исходные реагенты в отличие от пара-изомера, для которого скорость гидролиза мала.
Нитрозирование
Нитрозирование фенолов осуществляется с помощью азотистой кислоты в воде или уксусной кислоте. Эта реакция отличается очень высокой региоселективностью и замещение идет в пара-положение к гидроксильной группе. Типичное распределение орто- и пара-изомеров при нитрозировании можно проиллюстрировать на примере самого фенола.
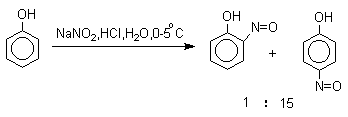
Алкилирование и ацилирование по Фриделю-Крафтсу
Так как фенолы взаимодействуют с галогенидами алюминия и другими кислотами Льюиса с образованием солей типа ArOAlCl2, прямое их алкилирование в условиях реакции Фриделя-Крафтса провести не удается. Фенолы алкилируют алкенами и спиртами в условиях кислотного катализа. В качестве катализаторов предпочитают использовать серную, фтористоводородную, фосфорную кислоты или катиониты КУ-2 и другие катионообменные смолы. Таким образом, из крезола и изобутилена в промышленности получают пространственно затрудненный фенол - 2,6-ди-трет-бутил-4-метилфенол (ионол), который широко применяется для стабилизации полимеров.
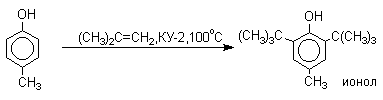
Аналогично из фенола и изопропилового спирта получается 2,4,6-триизопропилфенол.
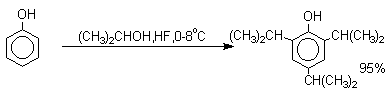
Ацилирование фенолов в классических условиях реакции Фриделя-Крафтса комплексом ацилгалогенида и хлорида алюминия приводит к неудовлетворительным результатам, так как ацилированию подвергается гидроксильная группа фенола. Более эффективна такая модификация этого метода, когда в качестве ацилирующего агента используется комплекс карбоновой кислоты и трехфтористого бора. Ацильная группа при этом вводится практически исключительно в пара-положение бензольного кольца. Так, например, фенол при взаимодействии с комплексом уксусной кислоты и BF3 дает пара-гидроксиацетофенон с 95%-ным выходом.
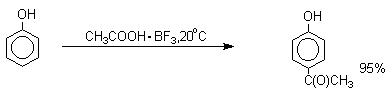
Наиболее общий метод получения гидроксикетонов ароматического ряда основан на перегруппировке Фриса. Ариловые эфиры карбоновых кислот при нагревании с AlCl3 или AlBr3 перегруппировываются в изомерные орто- и пара-гидроксикетоны.
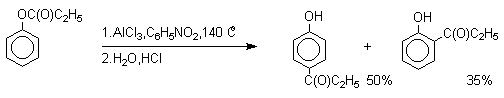
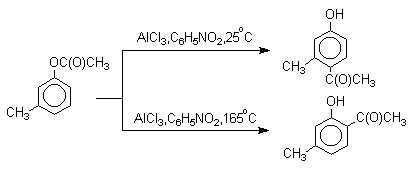
Соотношение орто- и пара-изомеров зависит главным образом от температуры и растворителя. В более жестких условиях преобладает орто-гидроксикетон, а при 20-25оС - пара-гидроксикетон.
Механизм перегруппировки Фриса, по-видимому, заключается в межмолекулярном ацилировании орто- или пара-положения бензольного кольца арилового эфира комплексом второй молекулы сложного эфира и AlCl3 с образованием ацильного производного гидроксикетона и фенола.
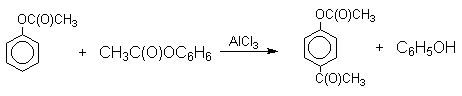
Перегруппировка завершается межмолекулярным переносом ацильной группы к фенолу.
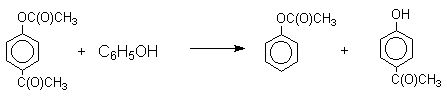
Формилирование
Формилирование – это введение группы СНО (см. лек.35). Синтетически наиболее важными являются формилирование фенолов по Вильсмейеру-Хааку и Реймеру-Тиману.
Реакция Вильсмейера-Хаака
N-Алкиламиды муравьиной кислоты - N,N-диметилформамид (ДМФА) и N-метилформамид в присутствии хлорокиси фосфора являются региоселективными формилирующими агентами. С помощью этих реагентов альдегидная группа вводится в пара-положение по отношению к ОН -группе. Эту реакцию можно также рассматривать как ацилирование, где роль катализатора (кислоты Льюиса) выполняют хлорокись фосфора (POCl3). Наиболее эффективна система ДМФА-POCl3, в которой ДМФА служит и реагентом, и растворителем. Электрофильным агентом в реакции Вильсмейера-Хаака является иминиевая соль, которая образуется при взаимодействии ДМФА и хлорокиси фосфора.
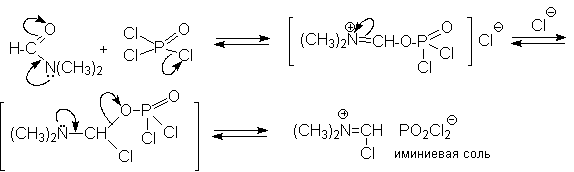
Иминиевая соль при необходимости может быть выделена в индивидуальном виде, однако обычно ее не выделяют и используют непосредственно после ее образования.
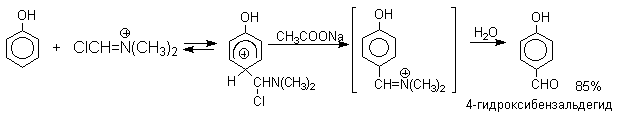
Реакция Вильсмейера-Хаака чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие выходы ароматических гидроксиальдегидов.
Реакция Реймера-Тимана
Формилирование фенолов по Реймеру-Тиману достигается при нагревании смеси фенола и большого избытка хлороформа с водным раствором гидроксида натрия при 50-70оС. Выходы альдегидов обычно невелики и редко превышают 30%, однако метод исключительно прост и доступен в практическом отношении. Главное достоинство реакции Реймера-Тимана заключается в преимущественном образовании орто-, а не пара-изомеров, как это имеет место в реакции Вильсмейера-Хаака.
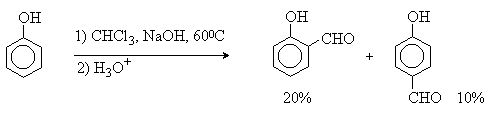
Механизм включает образование дихлоркарбена как интермедиата. Дихлоркарбен :CCl2 выполняет роль электрофильного агента по отношению к феноксид-иону, образующемуся в щелочной среде. Предполагаемый механизм реакции Реймера-Тимана может быть представлен следующей последовательностью превращений:
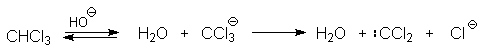
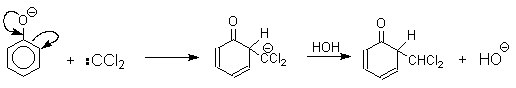
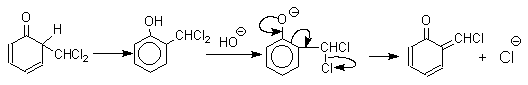
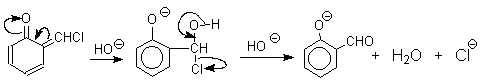
Реакция протекает только в сильно щелочной среде при наличии фенольного гидроксила, тогда как простые эфиры фенолов и диалкиланилины не формилируются в этих условиях.
Карбоксилирование феноксид-ионов (реакция Кольбе)
Будучи сильными нуклеофилами феноксид-анионы способны взаимодействовать с таким слабым электрофильным реагентом как оксид углерода (IV). При нагревании сухих фенолятов натрия или лития с СО2 при повышенном давлении, образуются натриевые или литиевые соли салициловой кислоты.
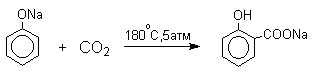
В аналогичных условиях из фенолятов калия, рубидия и цезия получаются только соли пара-гидроксибензойной кислоты.
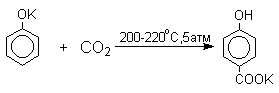
Такое различие в направлении карбоксилирования Na- и К-солей фенола принято объяснять различием в хелатообразовании этих двух катионов с атомом кислорода CO2 в переходном состоянии реакции, приводящем к салициловой кислоте. Катионы натрия и, особенно, лития значительно более эффективны по сравнению с катионом калия в способности к образованию координационной связи с атомом кислорода.
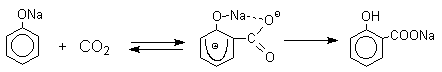
Предполагается, что для фенолятов калия, рубидия и цезия электрофильная атака осуществляется исключительно в пара-положение без какой-либо координации катиона по атому кислорода.
В отличие от одноатомных фенолов двухатомные и трехатомные фенолы карбоксилируются в более мягких условиях. Так, резорцин карбоксилируется при пропускании СО2 в водный раствор его дикалиевой соли при 50оС. При этом образуется 2,4-дигидроксибензойная кислота.
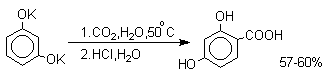
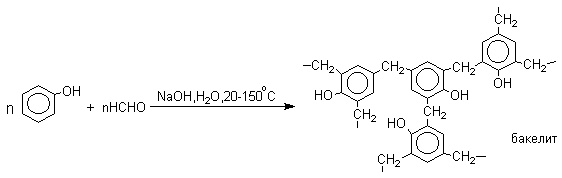
Конденсация с альдегидами и кетонами
Фенолы реагируют с формальдегидом в водном растворе в присутствии основания с образованием полимерного продукта, получившего название феноло-формальдегидной смолы, карболита или бакелита.
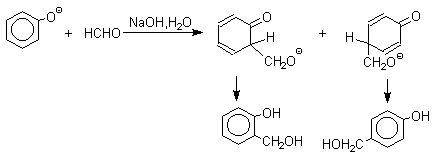
Взаимодействие феноксид-иона с формальдегидом напоминает альдольную конденсацию с той лишь разницей, что роль нуклеофильного агента вместо енолят-иона выполняет амбидентный феноксид-ион, а карбонильной компонентой является формальдегид.
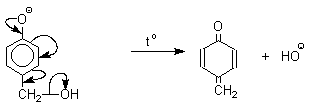
Подобно альдолям, орто- и пара-изомеры гидроксиметилфенола подвергаются дегидратации с образованием хинонметидов - соединений, родственных орто- и пара-хинонам.
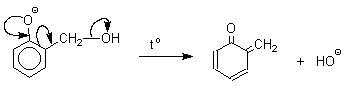
Последующее присоединение феноксид-иона к хинонметиду представляет собой присоединение амбидентного аниона к a ,b -непредельному кетону по Михаэлю.
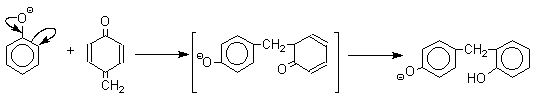
В результате дальнейшей поликонденсации в орто- и пара-положение к гидроксигруппе фенола получается трехмерная структура конечного продукта - бакелита. Бакелит представляет собой прозрачную смолу, в которой линейные звенья связаны "поперечными" связями в пара-положениях.
Фенол конденсируется с ацетоном в кислой среде с образованием так называемого бисфенола А.
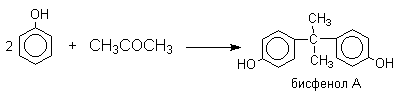
Получено много подобных продуктов конденсации фенолов с кетонами. Они находят применение в качестве антиоксидантов и мономеров для получения эпоксидных смол.
Окисление
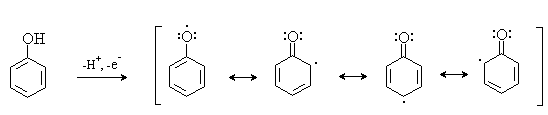
Окисление фенолов относится к числу сложных, многостадийных процессов, приводящих к продуктам разного строения. Механизм окисления может сильно меняться в зависимости от строения фенола и природы одно- или двухэлектронного окислителя. Механизм окисления включает стадию образования стабилизированных резонансом ароксильных радикалов.
Направление дальнейших превращений зависит от характера заместителей в ароматическом кольце. При окислении большинства фенолов образуется несколько различных форм димеров в результате образования новых связей С-С между орто-орто, орто-пара- и пара-пара-положениями ароксильных радикалов, а также новых С-О связей между атомом кислорода одного радикала и орто- или пара-положением другой радикальной частицы. Всего, таким образом, образуется потенциально не менее пяти различных типов димеров, которые находится в равновесии с исходным ароксильным радикалом. Например, для монозамещенного фенола:
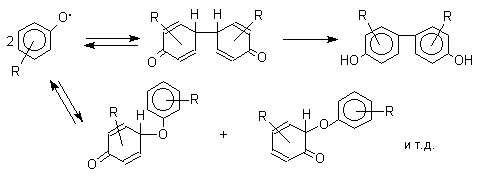
Димеры, возникающие в результате рекомбинации радикалов с образованием новой С-С связи, называются хинолидами. Хинолиды далее изомеризуются с образованием изомерных дигидроксибифенилов. Другой тип димеров, содержащих центральную связь С-О, носит название хиноловыхэфиров.
Ароксильные радикалы пространственно затрудненных фенолов, содержащие в обоих орто- и пара-положении третичные алкильные группы, мономерны и не проявляют тенденции к образованию димеров в растворе. Например, при окислении 2,4,6-три-трет-бутилфенола гексацианоферратом(III) калия K3Fe(CN)6 в бинарной системе бензол-вода в инертной атмосфере образуется устойчивый радикал одновалентного кислорода - три-трет-бутилфеноксил, окрашенный в синий цвет.
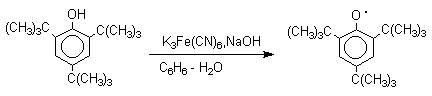
Этот радикал находится в мономерной форме в 0,1 молярном растворе в бензоле или эфире, а также в кристаллическом состоянии. Он очень чувствителен к действию кислорода воздуха, оксида азота (IY), оксида азота (II) и других радикальных частиц. Стабильность ароксильных радикалов пространственно затрудненых фенолов обусловливает их антиокислительные свойства. Они выполняют роль ловушек свободных радикалов в процессах пероксидного окисления. Активный свободный радикал, ведущий цепь окисления, быстро взаимодействует с таким фенолом, давая устойчивый ароксильный радикал, что приводит к обрыву цепи.
ArOH + RO2s® ArOs + ROOH
Пространственно затрудненные фенолы (ионол, гальваноксил) используют как антиоксиданты, стабилизирующие синтетические каучуки, пищевые жиры, витамины и др.